1.本发明属于光敏微晶玻璃技术领域,涉及光敏剂,具体涉及一种适用于光敏微晶玻璃的掺杂氧化铈光敏剂及其制备方法与应用。
背景技术:
2.随着电子工业的飞速发展,一种新型材料——li2o-al2o
3-sio2光敏微晶玻璃逐渐进入人们的视野。光敏微晶玻璃具有强度高、介电性能好等优点,可加工成各种形状的薄片,其优异的机械和电气性能使其在电子工业中具有巨大的应用潜力。相对较低的介电常数和介电损耗以及较低的成本使得光敏微晶玻璃有望取代硅材料,广泛应用于集成电路、生物医学、航空航天等领域。
3.氧化铈是li2o-al2o
3-sio2光敏微晶玻璃的重要组成部分,它可以作为捕获光子的光敏剂。其中ceo2是变价物质,可以在紫外线照射下释放自由电子,同时玻璃中的成核剂ag2o捕获自由电子后形成银原子。相关反应式如下所示:
4.ce
3
hv
→
ce
4
e-5.ag
e-→
ag
6.li2o-al2o
3-sio2光敏微晶玻璃经过上述曝光过程后再经过热处理,曝光部分玻璃中的银原子会聚集成银胶粒子,这些银胶粒子可以作为晶核诱导li2o
·
sio2的结晶。由于li2o
·
sio2晶相在hf溶液中的溶解速度比未曝光区域玻璃基质的快,因此可以通过上述步骤对光敏微晶玻璃表面或内部的微观结构进行加工。
7.虽然目前ceo2作为光敏微晶玻璃的光敏剂被广泛应用,但由于ceo2的吸收峰在320nm 左右,而目前工业上常用曝光光源为0.35um光刻机。ceo2吸收峰与工业曝光光源波段的失配导致光敏微晶玻璃的曝光效率和质量有所下降。目前对于光敏微晶玻璃生产来说,并没有波长为320nm的专用光刻机,如果为提高曝光效率和质量而制作波长为320nm的专用光刻机,则将极大提高生产成本。
技术实现要素:
8.针对现有光敏微晶玻璃的光敏剂ceo2存在的吸收峰与工业曝光光源波段的失配导致光敏微晶玻璃的曝光效率和质量低的问题,本发明的目的旨在提供一种适用于光敏微晶玻璃的掺杂氧化铈光敏剂及其制备方法与应用,通过自蔓延燃烧法对光敏剂ceo2进行掺杂改性,改善其光吸收性能使其吸收峰移动,以使其吸收峰与现有光源匹配,从而提高光敏微晶玻璃的成核效率与曝光质量。
9.为达到上述目的,本发明提供的一种适用于光敏微晶玻璃的掺杂氧化铈光敏剂,该掺杂氧化铈光敏由通式ce
1-x w
x
o2表示,通式中,x取值为:x=0.05~0.25。
10.上述适用于光敏微晶玻璃的掺杂氧化铈光敏剂,w掺杂量过大时会降低ceo2结晶度, x的取值优选为:x=0.1~0.2,进一步优选为x=0.1。
11.上述适用于光敏微晶玻璃的掺杂氧化铈光敏剂的制备方法,包括以下步骤:
12.(1)配料:以硝酸铈、偏钨酸铵水合物((nh4)6h2w
12o40
·
xh2o)以及有机燃料为原料,按通式ce
1-x w
x
o2中x的设定值确定的化学式进行称量配料,其中金属离子与有机燃料的摩尔比为1:1.5~2;
13.(2)溶解:将硝酸铈溶解于去离子水中,将偏钨酸铵水合物溶解于硝酸中,再将两种溶液混合形成混合溶液;
14.(3)制备前驱体溶液:将有机燃料加入到混合溶液中搅拌至溶解,在搅拌条件下加入氨水调节混合溶液ph值为7~8,获得前驱体溶液;
15.(4)制备前驱体粉末:将前驱体溶液加热至沸腾并持续搅拌,直至形成干凝胶,将干凝胶粉末点燃,发生自蔓延燃烧,得到疏松状的前驱体粉末;
16.(5)烧结:将得到的前驱体粉末进行烧结,烧结温度为950~1100℃,烧结时间为3~ 4h,自然冷却后得到烧结粉末;
17.(6)球磨:将冷却后的烧结粉末进行湿法球磨,将球磨后的浆料烘干后,得到化学式为 ce
1-x w
x
o2的掺杂氧化铈光敏剂。
18.上述适用于光敏微晶玻璃的掺杂氧化铈光敏剂的制备方法,步骤(1)中,金属离子指铈以及钨离子。
19.上述适用于光敏微晶玻璃的掺杂氧化铈光敏剂的制备方法,偏钨酸铵水合物溶解于硝酸中,对于硝酸的用量以及其浓度并没有特殊的要求,将偏钨酸铵水合物完全溶解即可,硝酸浓度采用常规市售硝酸溶液浓度或实验室常规浓度即可。进一步地,偏钨酸铵水合物中金属离子的摩尔量与硝酸的比例可以为:偏钨酸铵水合物中金属离子的摩尔量为0.01mol时,可将其溶解到浓度为14.4~15.2mol/l、体积为15ml的硝酸溶液中。在本发明的实施例中,硝酸溶液为市售购买,浓度为14.4~15.2mol/l。
20.上述适用于光敏微晶玻璃的掺杂氧化铈光敏剂的制备方法,有机燃料的作用是与硝酸盐在加热时发生强烈的氧化还原放热反应,以维持反应进程并引起自蔓延燃烧,因此有机燃料可以选择并不限于柠檬酸、氨基乙酸、草酸或聚丙烯酸等本领域常规有机燃料,进一步优选为柠檬酸,柠檬酸为三元弱酸,在水溶液中发生多级电离,然后再与金属离子发生络合反应。硝酸盐/柠檬酸的配比会影响凝胶的稳定性和自燃烧效果。
21.上述钬掺杂铁酸铜多铁陶瓷的制备方法中,体系的ph值会影响柠檬酸的多级电离,进而对溶胶中组分分布产生影响。反应体系ph值较低时,柠檬酸的多级电离收到抑制,溶胶体系中金属离子部分与柠檬酸根络合,部分仍以硝酸盐的形式存在,导致溶胶组分分布不均匀;当ph值较高时,柠檬酸的电离较完全,但碱性条件会使金属离子形成沉淀,不能充分被络合,同样影响凝胶组分的均匀性,进而影响干凝胶的自燃烧特性。本发明中,控制ph值为 7~8,在该范围内,可以实现柠檬酸根和金属离子的充分络合以及后续干凝胶的完全燃烧。
22.上述适用于光敏微晶玻璃的掺杂氧化铈光敏剂的制备方法,所述步骤(5)中,烧结升温速率采用常规速率即可,优选3~4℃/min。
23.上述适用于光敏微晶玻璃的掺杂氧化铈光敏剂的制备方法,所述步骤(6)中,湿法球磨可以采用本领域常规的球磨介质和分散剂,以及常规球磨参数,并无特殊限定。在本发明中,优选以乙醇为介质、三乙醇胺为分散剂,对烧结粉末进行湿法球磨。
24.本发明提供的适用于光敏微晶玻璃的掺杂氧化铈光敏剂及其制备方法,其掺杂原理是: ceo2的带隙约为3.2ev,这源于氧的2p上的电子在向ce的4f上的跃迁,掺杂后的ceo2纳米颗粒,由于掺杂金属w提供比4f(ce)低的未占据轨道,电子从2p(o)轨道上跃迁到未占据的轨道需要的能量较低,所以掺杂后的ceo2纳米颗粒比未掺杂的ceo2能吸收更多的波长较长的光,导致其吸收向长波方向移动,产生红移现象。
25.本发明还提供了上述适用于光敏微晶玻璃的掺杂氧化铈光敏剂作为光敏剂在光敏微晶玻璃中的应用。
26.与现有技术相较,本发明提供的技术方案具有以下有益效果:
27.(1)本发明提供的适用于光敏微晶玻璃的掺杂氧化铈光敏剂,对现有光敏剂ceo2进行 w离子掺杂,掺杂后的ceo2纳米颗粒,由于掺杂金属w提供比4f(ce)低的未占据轨道,电子从2p(o)轨道上跃迁到未占据的轨道需要的能量较低,因而掺杂后的ceo2纳米颗粒比未掺杂的ceo2能吸收更多的波长较长的光,导致其吸收向长波方向移动,产生红移现象,由此改善其光吸收性能使其吸收峰移动,使其吸收峰与现有光源匹配,作为光敏剂应用于光敏微晶玻璃中,可以有效提高光敏微晶玻璃的成核效率与曝光质量。
28.(2)本发明提供的适用于光敏微晶玻璃的掺杂氧化铈光敏剂的制备方法,利用自蔓延燃烧法合成掺杂氧化铈光敏剂,无需预烧,烧结成相温度低,烧结时间短,达到了节能的良好效果;且制备工艺简单,制备所用到的设备价格低廉,成本低,制备工艺周期短,具有良好的应用前景,值得在本领域推广应用。
附图说明
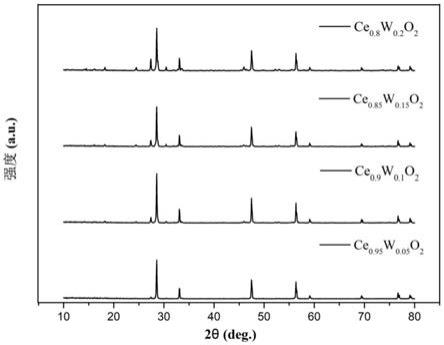
29.图1是实施例1-4制备的掺杂不同浓度w的氧化铈光敏剂样品的xrd图谱;
30.图2是实施例2制备的掺杂w的氧化铈光敏剂样品以及对纯的ceo2光敏剂样品的紫外
ꢀ‑
可见吸收光谱,其中图(a)为200nm~650nm波段的吸收光谱,图(b)为图(a)局部图,即300nm~400nm波段的吸收光谱;
31.图3是应用例中ag2co3和ceo2样品的ag 3d和ce 3d xps图谱,其中图(a)和图(b) 分别为未曝光的ag 3d和ce 3d xps图谱,图(c)和图(d)分别为经320nm紫外光光源曝光的ag 3d和ce 3d xps图谱,图(e)和图(f)分别为经350nm紫外光光源曝光的ag 3d 和ce 3d xps图谱;
32.图4是应用例中ag2co3和ceo2样品的ag 3d和ce 3d xps图谱,其中图(a)和图(b) 分别为未曝光的ag 3d和ce 3d xps图谱,图(c)和图(d)分别为经320nm紫外光光源曝光的ag 3d和ce 3d xps图谱,图(e)和图(f)分别为经350nm紫外光光源曝光的ag 3d 和ce 3d xps图谱。
具体实施方式
33.以将结合附图对本发明各实施例的技术方案进行清楚、完整的描述,显然,所描述实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施例,都属于本发明。
34.实施例1
ce
0.85w0.15
o2确定的化学式进行称量配料,其中金属离子与有机燃料的摩尔比为1:1.5;
53.(2)溶解:将0.0255mol ce(no3)3·
6h2o溶解于去离子水中,将0.000375mol(nh4) 6
h2w
12o40
·
xh2o溶解于15ml浓度为14.4-15.2mol/l的硝酸中,再将两种溶液混合形成混合溶液;
54.(3)制备前驱体溶液:将0.045mol柠檬酸加入到混合溶液中搅拌至完全溶解,在搅拌条件下缓慢加入30ml氨水,观察到絮状沉淀出现,然后消失,溶液最终澄清的现象,调节ph 至7~8,获得前驱体溶液;
55.(4)制备前驱体粉末:将前驱体溶液转移至坩埚中,于电炉上加热至沸腾并持续搅拌,直至形成干凝胶,将干凝胶粉末点燃,发生自蔓延燃烧,得到疏松状的前驱体粉末;
56.(5)烧结:将得到的前驱体粉末收集于小坩埚中,放入马弗炉进行烧结,烧结温度为 1000℃,烧结时间为3h,自然冷却后得到烧结粉末;
57.(6)球磨:收集冷却后的烧结粉末,以乙醇为介质,三乙醇胺为分散剂,进行湿法球磨,将球磨后的浆料50℃下在烘箱中烘干后,得到化学式为ce
0.85w0.15
o2的掺杂氧化铈光敏剂。
58.实施例4
59.本实施例利用柠檬酸-硝酸盐自蔓延燃烧法,制备由化学式ce
0.8w0.2
o2(即x=0.2)表示的适用于光敏微晶玻璃的掺杂氧化铈光敏剂,具体包括以下步骤:
60.(1)配料:以ce(no3)3·
6h2o、(nh4)6h2w
12o40
·
xh2o以及柠檬酸为原料,按化学式 ce
0.8w0.2
o2确定的化学式进行称量配料,其中金属离子与有机燃料的摩尔比为1:1.5;
61.(2)溶解:将0.024mol ce(no3)3·
6h2o溶解于去离子水中,将0.0005mol(nh4) 6
h2w
12o40
·
xh2o溶解于15ml浓度为14.4-15.2mol/l的硝酸中,再将两种溶液混合形成混合溶液;
62.(3)制备前驱体溶液:将0.045mol柠檬酸加入到混合溶液中搅拌至完全溶解,在搅拌条件下缓慢加入30ml氨水,观察到絮状沉淀出现,然后消失,溶液最终澄清的现象,调节ph 至7~8,获得前驱体溶液;
63.(4)制备前驱体粉末:将前驱体溶液转移至坩埚中,于电炉上加热至沸腾并持续搅拌,直至形成干凝胶,将干凝胶粉末点燃,发生自蔓延燃烧,得到疏松状的前驱体粉末;
64.(5)烧结:将得到的前驱体粉末收集于小坩埚中,放入马弗炉进行烧结,烧结温度为 1000℃,烧结时间为3h,自然冷却后得到烧结粉末;
65.(6)球磨:收集冷却后的烧结粉末,以乙醇为介质,三乙醇胺为分散剂,进行湿法球磨,将球磨后的浆料50℃下在烘箱中烘干后,得到化学式为ce
0.8w0.2
o2的掺杂氧化铈光敏剂。
66.实施例5
67.本实施例利用柠檬酸-硝酸盐自蔓延燃烧法,制备由化学式ce
0.75w0.25
o2(即x=0.25)表示的适用于光敏微晶玻璃的掺杂氧化铈光敏剂,具体包括以下步骤:
68.(1)配料:以ce(no3)3·
6h2o、(nh4)6h2w
12o40
·
xh2o以及柠檬酸为原料,按化学式 ce
0.75w0.25
o2确定的化学式进行称量配料,其中金属离子与有机燃料的摩尔比为1:1.5;
69.(2)溶解:将0.0225mol ce(no3)3·
6h2o溶解于去离子水中,将0.000625mol(nh4) 6
h2w
12o40
·
xh2o溶解于15ml浓度为14.4-15.2mol/l的硝酸中,再将两种溶液混合形成混合
溶液;
70.(3)制备前驱体溶液:将0.045mol柠檬酸加入到混合溶液中搅拌至完全溶解,在搅拌条件下缓慢加入30ml氨水,观察到絮状沉淀出现,然后消失,溶液最终澄清的现象,调节ph 至7~8,获得前驱体溶液;
71.(4)制备前驱体粉末:将前驱体溶液转移至坩埚中,于电炉上加热至沸腾并持续搅拌,直至形成干凝胶,将干凝胶粉末点燃,发生自蔓延燃烧,得到疏松状的前驱体粉末;
72.(5)烧结:将得到的前驱体粉末收集于小坩埚中,放入马弗炉进行烧结,烧结温度为 1000℃,烧结时间为3h,自然冷却后得到烧结粉末;
73.(6)球磨:收集冷却后的烧结粉末,以乙醇为介质,三乙醇胺为分散剂,进行湿法球磨,将球磨后的浆料50℃下在烘箱中烘干后,得到化学式为ce
0.75w0.25
o2的掺杂氧化铈光敏剂。
74.实施例6
75.本实施例利用柠檬酸-硝酸盐自蔓延燃烧法,制备由化学式ce
0.9w0.1
o2(即x=0.1)表示的适用于光敏微晶玻璃的掺杂氧化铈光敏剂,具体包括以下步骤:
76.(1)配料:以ce(no3)3·
6h2o、(nh4)6h2w
12o40
·
xh2o以及柠檬酸为原料,按化学式 ce
0.9w0.1
o2确定的化学式进行称量配料,其中金属离子与有机燃料的摩尔比为1:2;
77.(2)溶解:将0.027mol ce(no3)3·
6h2o溶解于去离子水中,将0.00025mol(nh4) 6
h2w
12o40
·
xh2o溶解于15ml浓度为14.4-15.2mol/l的硝酸中,再将两种溶液混合形成混合溶液;
78.(3)制备前驱体溶液:将0.06mol柠檬酸加入到混合溶液中搅拌至完全溶解,在搅拌条件下缓慢加入30ml氨水,观察到絮状沉淀出现,然后消失,溶液最终澄清的现象,调节ph 至7~8,获得前驱体溶液;
79.(4)制备前驱体粉末:将前驱体溶液转移至坩埚中,于电炉上加热至沸腾并持续搅拌,直至形成干凝胶,将干凝胶粉末点燃,发生自蔓延燃烧,得到疏松状的前驱体粉末;
80.(5)烧结:将得到的前驱体粉末收集于小坩埚中,放入马弗炉进行烧结,烧结温度为950℃,烧结时间为3h,自然冷却后得到烧结粉末;
81.(6)球磨:收集冷却后的烧结粉末,以乙醇为介质,三乙醇胺为分散剂,进行湿法球磨,将球磨后的浆料50℃下在烘箱中烘干后,得到化学式为ce
0.9w0.1
o2的掺杂氧化铈光敏剂。
82.实施例7
83.本实施例利用柠檬酸-硝酸盐自蔓延燃烧法,制备由化学式ce
0.9w0.1
o2(即x=0.1)表示的适用于光敏微晶玻璃的掺杂氧化铈光敏剂,具体包括以下步骤:
84.(1)配料:以ce(no3)3·
6h2o、(nh4)6h2w
12o40
·
xh2o以及柠檬酸为原料,按化学式 ce
0.9w0.1
o2确定的化学式进行称量配料,其中金属离子与有机燃料的摩尔比为1:2;
85.(2)溶解:将0.027mol ce(no3)3·
6h2o溶解于去离子水中,将0.00025mol(nh4) 6
h2w
12o40
·
xh2o溶解于15ml浓度为14.4-15.2mol/l的硝酸中,再将两种溶液混合形成混合溶液;
86.(3)制备前驱体溶液:将0.06mol柠檬酸加入到混合溶液中搅拌至完全溶解,在搅拌条件下缓慢加入30ml氨水,观察到絮状沉淀出现,然后消失,溶液最终澄清的现象,调节
ph 至7~8,获得前驱体溶液;
87.(4)制备前驱体粉末:将前驱体溶液转移至坩埚中,于电炉上加热至沸腾并持续搅拌,直至形成干凝胶,将干凝胶粉末点燃,发生自蔓延燃烧,得到疏松状的前驱体粉末;
88.(5)烧结:将得到的前驱体粉末收集于小坩埚中,放入马弗炉进行烧结,烧结温度为 1100℃,烧结时间为3h,自然冷却后得到烧结粉末;
89.(6)球磨:收集冷却后的烧结粉末,以乙醇为介质,三乙醇胺为分散剂,进行湿法球磨,将球磨后的浆料50℃下在烘箱中烘干后,得到化学式为ce
0.9w0.1
o2的掺杂氧化铈光敏剂。
90.对比例
91.本对比例利用柠檬酸-硝酸盐自蔓延燃烧法,制备光敏微晶玻璃现有光敏剂ceo2,具体包括以下步骤:
92.(1)配料:以ce(no3)3·
6h2o以及柠檬酸为原料,进行称量配料,其中金属离子与有机燃料的摩尔比为1:1.5;
93.(2)溶解:将0.03mol ce(no3)3·
6h2o溶解于去离子水中形成硝酸铈溶液;
94.(3)制备前驱体溶液:将0.045mol柠檬酸加入到硝酸铈溶液中搅拌至完全溶解,在搅拌条件下缓慢加入30ml氨水,观察到絮状沉淀出现,然后消失,溶液最终澄清的现象,调节 ph至7~8,获得前驱体溶液;
95.(4)制备前驱体粉末:将前驱体溶液转移至坩埚中,于电炉上加热至沸腾并持续搅拌,直至形成干凝胶,将干凝胶粉末点燃,发生自蔓延燃烧,得到疏松状的前驱体粉末;
96.(5)烧结:将得到的前驱体粉末收集于小坩埚中,放入马弗炉进行烧结,烧结温度为 1000℃,烧结时间为3h,自然冷却后得到烧结粉末;
97.(6)球磨:收集冷却后的烧结粉末,以乙醇为介质,三乙醇胺为分散剂,进行湿法球磨,将球磨后的浆料50℃下在烘箱中烘干后,得到化学式为ceo2光敏剂。
98.以下对实施例1-5制备的掺杂氧化铈光敏剂样品以及对比例制备的ceo2光敏剂样品进行性能分析。
99.(一)结构分析
100.为了研究通过本发明方法制备的掺杂氧化铈光敏剂样品的相结构,采用x射线衍射仪 (xrd)对实施例1-4制备的掺杂氧化铈光敏剂样品进行物相分析,结果见图1所示。从图1中可以看出,所有样品均出现了和立方萤石结构ceo2的jcpds卡片相吻合的衍射峰,没有原材料及其它杂质的衍射峰,说明所有样品均为单相结构,掺杂离子并没有改变基体的结构。
101.(二)紫外吸收分析
102.为了研究通过本发明方法制备的掺杂氧化铈光敏剂样品的紫外-可见光吸收性能,对实施例2制备的掺杂氧化铈光敏剂样品以及对比例制备的ceo2光敏剂样品进行紫外吸收分析,结果如图2所示。从图2可以看出,在250nm~320nm波段,ceo2的吸收强度最高;ce
0.9w0.1
o2样品在350nm波段附近和之后的吸收强度最高。
103.应用例
104.将实施例2制备的ce
0.9w0.1
o2和ag2co3按质量比值为0.09的比例称取60g的粉料,将对比例制备的ceo2和ag2co3粉末按质量比值为0.08的比例称取60g的粉料,分别在研钵中力
度适中的研磨1小时,称取20克混合均匀的粉料在320nm的光源下曝光30分钟,称取20 克混合均匀的粉料在350nm的光源下曝光30分钟,剩余20克粉料不曝光。对曝光和未曝光的样品均进行xps测试,测试结果如图3和图4所示。
105.从图3可以看出,ag2co3和ceo2样品的xps图谱在未曝光前只有ag
的峰,表明没有单质ag的存在。在经过320nm或350nm紫外光源曝光后,都出现了ag0的峰值,表明有单质ag的析出。与此同时,经过320nm或350nm紫外光源曝光后曝光ce
3
的峰面积都比未曝光的小,表明部分ce
3
经曝光转变为ce
4
。同时可以看到ag2co3和ceo2样品在320nm紫外光源下曝光后ag0的峰面积最大,ce
3
的峰面积最小,表明ceo2在320nm条件下曝光性能最佳。
106.从图4可以看出,ag2co3和ce
0.9w0.1
o2的xps图谱在曝光前有ag
峰,几乎没有ag0峰。在320nm或350nm紫外光曝光后,明显出现ag0的峰,说明ag单质的析出。与未曝光的样品相比,在320nm紫外光源曝光后的样品的ce
3
峰面积略有下降,而在350nm紫外光源曝光后的样品的ce
3
峰面积显著降低,表明ag2co3和ce
0.9w0.1
o2的样品在350nm条件下会有为更多的ce
3
转换为ce
4
。在350nm紫外光光源下曝光后,样品的ag0的峰面积最大, ce
3
的峰面积最小,说明样品ce
0.9w0.1
o2在350nm处的曝光性能最好。
107.本领域的普通技术人员将会意识到,这里所述的实施例是为了帮助读者理解本发明的原理,应被理解为本发明的保护范围并不局限于这样的特别陈述和实施例。本领域的普通技术人员可以根据本发明公开的这些技术启示做出各种不脱离本发明实质的其它各种具体变形和组合,这些变形和组合仍然在本发明的保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。