1.本发明涉及一种联苯双酯的改进合成方法,属于医药中间体技术领域。
背景技术:
2.联苯双酯(全称4,4-二甲氧基-5,6,5',6'-双(亚甲二氧基)联苯-2,2'-二羟酸二甲酯),白色晶状固体,是治疗病毒性肝炎和药物性肝损伤引起转氨酶升高的常用药物。具有保护肝细胞,增加肝脏的解毒功能的药理作用,尤其是其降酶作用,效果明显,且毒性低,副作用小。
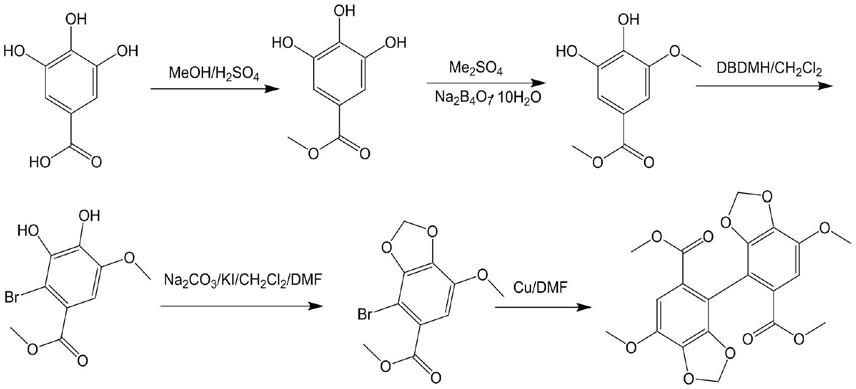
3.目前,联苯双酯的合成工艺主要为采用没食子酸为原料,经过五步反应后得到联苯双酯,详细工艺如下:
[0004][0005]
第一步,以没食子酸为原料,在甲醇/浓硫酸体系中,将羧酸转变为酯化物;第二步,酯化物在硼砂水体系中滴加硫酸二甲酯将羟基成单甲醚;第三步,单甲醚在二氯甲烷溶剂与二溴海因溴化反应得到溴代物;第四步,溴代物在碳酸钠/碘化钾/dmf/二氯甲烷体系中,将其它两个羟基生成环化物;第五步,环化物在dmf/铜粉体系中偶联,得到联苯双酯。
[0006]
该反应体系中,涉及反应步骤较多,后处理相对较为繁琐,因而有必要对现有合成工艺进行改进。
技术实现要素:
[0007]
为了克服上述技术缺陷,本发明公开了一种联苯双酯的改进合成方法。以没食子酸为原料,在碱性条件下,与二卤代物反应生成环化物,接着与硫酸二甲酯反应,羧酸成酯的同时,羟基成醚;最后采用大位阻碱去质子,在无水三氯化铁存在下偶联,得到联苯双酯。该方法仅有三步反应,最后一步反应时,偶联生成的异构体,采用甲基叔丁基醚/乙醇体系,打浆后去除,得到高纯产品联苯双酯。
[0008]
本发明所述一种联苯双酯的改进合成方法,采用方程式表示如下:
[0009]
包括如下步骤:
[0010]
第一步,环化反应
[0011]
以没食子酸为原料,在催化剂和碱存在下,与二卤代物反应生成环化物;
[0012]
第二步,酯化和醚化反应
[0013]
环化物与硫酸二甲酯反应,羧酸成酯/羟基成醚,得到酯化/醚化物;
[0014]
第三步,偶联反应
[0015]
酯化/醚化物采用大位阻碱去质子,在无水三氯化铁存在下偶联,得到联苯双酯。
[0016]
进一步地,在上述技术方案中,第一步所述碱选自碳酸钠或碳酸钾;反应在环丁砜溶剂中进行。
[0017]
进一步地,在上述技术方案中,第一步所述催化剂选自碘化钠或碘化钾;与四丁基碘化铵组成的混合催化体系。
[0018]
进一步地,在上述技术方案中,第一步所述二卤代物选自二氯甲烷、氯溴甲烷、二溴甲烷或二碘甲烷。
[0019]
进一步地,在上述技术方案中,第一步所述碱、二卤代物与没食子酸摩尔比为4-6:1.2-2.0:1;催化剂加入量为没食子酸重量的2-6%。
[0020]
进一步地,在上述技术方案中,第二步所述硫酸二甲酯与环化物摩尔比为2-4:1。
[0021]
进一步地,在上述技术方案中,第三步所述大位阻碱选自2,2,6,6-四甲基哌啶锂(templi);采用二异丙基乙基胺(lda)时,副产物比例较大。
[0022]
进一步地,在上述技术方案中,第三步所述无水三氯化铁、大位阻碱与酯化/醚化物摩尔比为1.2-1.8:1-1.2:1。采用无水三氯化铁偶联通常在3-4天(原料仍有5%以内剩余),加入碘化亚铜催化反应时,可以缩短反应时间(原料仍有5%以内剩余)。反应过程中存在一定比例的副产物,结构为:采用甲基叔丁基醚/乙醇体系,打浆后可将该副产物去除,本步反应收率在55-63%。
[0023]
发明有益效果
[0024]
本发明以没食子酸为原料,经过三步反应即可完成,避免了传统工艺中五步反应,缩短了反应步骤,合成工艺适合工业化放大。采用的原料和反应试剂均为市场易得,针对工艺中的难点,最后一步去质子偶联时,存在一定量副产物和剩余原料的情况,经过条件筛选,找到混合溶剂打浆的方式去除,避免了柱层析纯化。
具体实施例
[0025]
实施例1
[0026]
第一步:
[0027]
在三口瓶中加入环丁砜250ml、二溴甲烷(24.4g,0.14mol)、碳酸钾(69.0g,0.50mol)、碘化钾(3.2g)和四丁基碘化铵(1.5g),搅拌均匀后,升温至75-80℃形成均匀混合溶液。控温70-80℃滴加没食子酸(17.0g,0.1mol)/环丁砜55ml溶液,滴加完毕,搅拌升温至95-100℃保温6小时,tlc检测原料反应完毕。
[0028]
反应液降至室温过滤,固体再次环丁砜(60ml)打浆,滤液加入10%盐酸调至ph=2-3,然后加入2.4l庚烷溶剂后搅拌,析出的固体过滤。固体加入二氯甲烷溶解,二氯甲烷层采2%盐酸水溶液洗两次,合并有机层,无水硫酸镁干燥。过滤,减压蒸干后采用甲苯溶剂打浆得到浅灰色固体15.3g,收率84%。hnmr(cd3od,400mhz):7.22(s,1h),7.16(s,1h),6.44(s,2h).
[0029]
第二步:
[0030]
在三口瓶中,加入环化物(18.4g,0.1mol),dmf(60ml)和硫酸二甲酯(12.6g,0.1mol),升温至120℃反应5小时,tlc检测酯化完毕,接着加入水144克,降温至30-35℃搅拌均匀。控温不超过35℃滴加5%氢氧化钠水溶液调ph=9-10,然后滴加硫酸二甲酯(15.1g,0.12mol),同时滴加5%氢氧化钠水溶液维持反应液ph=9-10,整个过程耗时40分钟。随后30-35℃保温4小时,tlc原料反应完毕。
[0031]
降至室温,反应液加入二氯甲烷萃取四次(80ml*4),合并有机层,水洗两次,饱和食盐水洗两次,无水硫酸镁干燥,旋蒸掉有机溶剂,加入甲苯溶剂重结晶,降温过滤,得到土黄色固体酯化/醚化物19.5g,收率93%。hnmr(cdcl3,400mhz):7.30(s,1h),7.21(s,1h),6.06(s,2h),3.93(s,3h),3.88(s,3h).
[0032]
第三步:
[0033]
氮气保护下,在三口瓶内加入2,2,6,6-四甲基哌啶(31.0g,0.22mol)和四氢呋喃120ml,冷却至-45℃至-40℃,开始滴加n-buli(1.6m,145ml),滴毕保持温度在-45℃至-40℃搅拌30分钟。接着控温不超过-10℃缓慢滴加酯化/醚化物42.0g/160ml四氢呋喃溶液,滴毕保持温度在-50℃至-40℃搅拌4小时,形成溶液a待用;
[0034]
氮气保护下,在配有机械搅拌三口瓶内,控温-70℃至-60℃,将无水三氯化铁(52.0g,0.32mol)和碘化亚铜(6.3g)分散在160ml无水四氢呋喃中,形成较均匀悬浮溶液。接着将上述溶液a加入该悬浮溶液中,密闭反应22小时,后期tlc显示尚有少量原料剩余(且不再进行,hplc显示产物/副产物/原料归一化面积比为84/12/4,同样条件下,采用lda做碱时,三者比例为63/32/5)。
[0035]
将上述反应体系过滤,固体再次加入四氢呋喃450ml*2混洗均匀后,再次过滤,滤液旋干溶剂后,加入600ml二氯甲烷溶解,依次用10%盐酸水溶液、5%盐酸水溶液,饱和食盐水洗,无水硫酸镁干燥。过滤旋至溶剂为210ml左右,加热使固体全部溶解后,放置自然结晶,过滤得到固体粗品(颜色很黑),加入甲基叔丁基醚和乙醇混合溶剂1/5打浆,得到白色晶体26.0g,收率62%,hplc:99.7%。hnmr(cdcl3,400mhz):7.40(s,2h),6.01(s,4h),3.99(s,6h),3.69(s,6h).
[0036]
实施例2
[0037]
第一步:
[0038]
在三口瓶中加入环丁砜220ml、二溴甲烷(24.4g,0.14mol)、碳酸钠(53.0g,
0.50mol)、碘化钾(1.3g)和四丁基碘化铵(3.5g),搅拌均匀后,升温至75-80℃形成均匀混合溶液。控温70-80℃滴加没食子酸(17.0g,0.1mol)/环丁砜55ml溶液,滴加完毕,搅拌升温至95-100℃保温6小时,tlc检测原料反应完毕。
[0039]
反应液降至室温过滤,固体再次环丁砜(60ml)打浆,滤液加入10%盐酸调至ph=2-3,然后加入2.4l庚烷溶剂后搅拌,析出的固体过滤。固体加入二氯甲烷溶解,二氯甲烷层采2%盐酸水溶液洗两次,合并有机层,无水硫酸镁干燥。过滤,减压蒸干后采用甲苯溶剂打浆得到浅灰色固体15.7g,收率86%。
[0040]
第二步:
[0041]
在三口瓶中,加入环化物(18.4g,0.1mol),dmf(60ml)和硫酸二甲酯(12.6g,0.1mol),升温至120℃反应5小时,tlc检测酯化完毕,接着加入水144克,降温至30-35℃搅拌均匀。控温不超过35℃滴加5%氢氧化钠水溶液调ph=9-10,然后滴加硫酸二甲酯(15.1g,0.12mol),同时滴加5%氢氧化钠水溶液维持反应液ph=9-10,整个过程耗时40分钟。随后30-35℃保温4小时,tlc原料反应完毕。
[0042]
降至室温,反应液加入二氯甲烷萃取四次(80ml*4),合并有机层,水洗两次,饱和食盐水洗两次,无水硫酸镁干燥,旋蒸掉有机溶剂,加入甲苯溶剂重结晶,降温过滤,得到土黄色固体酯化/醚化物19.5g,收率93%。
[0043]
第三步:
[0044]
氮气保护下,在三口瓶内加入2,2,6,6-四甲基哌啶(31.0g,0.22mol)和四氢呋喃120ml,冷却至-40℃至-45℃,开始滴加n-buli(2.5m,96ml),滴毕保持温度在-45℃至-40℃搅拌30分钟。接着控温不超过-10℃缓慢滴加酯化/醚化物42.0g/160ml四氢呋喃溶液,滴毕保持温度在-50℃至-40℃搅拌4小时,形成溶液a待用;
[0045]
氮气保护下,在配有机械搅拌三口瓶内,控温-70℃至-60℃,将无水三氯化铁(38.9g,0.24mol)和碘化亚铜(4.4g)分散在160ml无水四氢呋喃中,形成较均匀悬浮溶液。接着将上述溶液a加入该悬浮溶液中,密闭反应25小时,后期tlc显示尚有少量原料剩余(且不再进行,hplc显示产物/副产物/原料归一化面积比为82/13/5)。
[0046]
将上述反应体系过滤,固体再次加入四氢呋喃450ml*2混洗均匀后,再次过滤,滤液旋干溶剂后,加入600ml二氯甲烷溶解,依次用10%盐酸水溶液、5%盐酸水溶液,饱和食盐水洗,无水硫酸镁干燥。过滤旋至溶剂为220ml左右,加热使固体全部溶解后,放置自然结晶,过滤得到固体粗品(颜色很黑),加入甲基叔丁基醚和乙醇混合溶剂1/5打浆,得到白色晶体24.8g,收率59%,hplc:99.2%。
[0047]
以上实施例描述了本发明的基本原理、主要特征及优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。