1.本发明涉及乙烯聚合催化剂领域,具体涉及用于制备超高分子量聚乙烯粉末的催化剂前体及催化剂。
背景技术:
2.超高分子量聚乙烯(uhmwpe)是一种相对分子质量极大的线型结构聚乙烯,由于具有异于通用聚乙烯的诸多优异性能而被广泛研究与应用。uhmwpe纤维是将uhmwpe原料通过凝胶纺丝法制备出的一种高性能纤维,其强度、模量高,机械性能优异,已在军工、国防等领域得到广泛应用。一般来说,uhmwpe粉料的相对分子质量分布、颗粒形态、粒径分布等性能参数会受到催化剂、聚合工艺等因素的影响,而这些性能参数又会影响聚合物粉料的可加工性,进而影响产品性能。因此,为了得到性能优异的uhmwpe纤维,需要从uhmwpe原料及纤维制备这两个方面进行探究。要制备超强聚乙烯纤维就对uhmwpe有着特殊要求,除了分子量要高于400万外,还要求催化活性高,这样所得uhmwpe的灰粉更低;同时所得聚乙烯的颗粒堆密度高;分布要均匀,粒径分布大于0.8;粒径不能太大也不能太小,平均粒径通常在80-120μm。
3.超高分子量聚乙烯粉末通常熔融时的粘度高,难以利用注射成型等进行加工,因此多溶解于溶剂中进行成型。特别是作为高强度纤维,例如,将使超高分子量聚乙烯粉末悬浮在溶剂中或者在加热下使超高分子量聚乙烯粉末溶胀-溶解在溶剂中而得到的物质向螺杄式挤岀机进料,从模头的喷丝头挤岀、拉伸,由此得到超高分子量聚乙烯纤维。此时,超高分子量聚乙烯粉末在溶剂中的溶解性低的情况下,会产生粉末的溶解残留,导致挤出机的筛网堵塞。即使能够纺丝,也会引起未熔融成分成为断裂点而在拉伸时产生断丝这样的问题。特别是纤维的情况下,表现出更高强度时需要更高度的拉伸,因此粉末的溶解残留导致生产时的断丝的缺陷,成为重大问题。
4.近年来,为了纤维的更高强度化、由纤维制造的产品的更轻量化,实施了使用具有更高粘均分子量的超高分子量聚乙烯,但是溶解残留所导致的不良情况经常发生这样的问题变得显著。
5.cn101245116a提供了一种用于制备超高分子量聚乙烯的催化体系。这个催化体系可以通过加入外给电子体,来调节聚乙烯的分子量。但聚乙烯的堆密度不高,且粒径在160μm左右,还不能满足纺丝需求。同时,采用外给电子体调节分子量的方法,比较麻烦,不能适应工业化生产的需求。
6.cn200410024103.8公开了一种超高分子量聚乙烯的制备方法,通过该方法制备的超高分子量聚乙烯具有良好的流动速率和加工性能。但是由于该发明中的超高分子量聚乙烯树脂分子量低,力学性能较低,不适用于高强高模量的纤维的生产。
7.cn1106025a公开了一种具有高堆积密度的超高分子量聚乙烯的制备方法,采用汽油作为溶剂,提供了一种具有高堆积密度的超高分子量聚乙烯,堆积密度范围在350-460
克/升。但是采用汽油作为溶剂,超高分子量聚乙烯的质量不稳定,力学性能较差,不利于制得高强度的聚乙烯纤维。另外,由于汽油馏程宽,能耗和成本高,不利于工业化生产。
8.cn1033703c报道了一种制备uhmwpe的方法,主要在催化剂体系中添加zncl2组分,通过控制zn/ti摩尔比可在60-610万之间调节uhmwpe的分子量。用此催化剂可获得良好粒子分布的uhmwpe。但此催化剂制备比较烦琐,要通过硏磨法制备,要制备不同分子量的uhmwpe就需要调节zn/ti摩尔比而制备不同的催化剂,且该体系催化活性较低。
9.cn1189486c提供了一种制备高堆密度、良好粒子形态的uhmwpe的催化体系,该催化剂通过卤化镁和铝化合物在醇存在下制备了镁-铝溶液,然后再与给电子体作用后加入钛化合物和硅化合物从而制备出催化剂,尽管该催化体系具有较好的催化活性,但所得超高分子量聚乙烯堆密度不够高,最大只有0.40g/cm3,粒径在152-179μm,且粒径分布比较宽,粒径分布指数在0.6左右。这种形态的聚乙烯粉末不利于纺丝。
10.cn1746197a提供了一种制备高堆密度的uhmwpe的催化体系,这种催化剂采用先制备载体然后载钛形成催化剂,从中可以看出其催化剂制备是比较烦琐的。在制备uhmwpe中添加了所谓性能调节剂有杋硅化合物。虽然所制备的uhmwpe具有较高的分子量,但没有提供聚合物粒子的平均粒径,且催化活性也不是很高,聚合时间为4小时其催化活性也只有3万倍左右。
11.目前制备uhmwpe不仅要求催化剂有较高的催化活性,而且具有动力学稳定和长效性,这样可以最大限度防止聚合物出现过大颗粒或过小颗粒并减少聚合物的灰份,这点尤其在制备超强聚乙烯纤维和锂电池隔膜更为重要。同时,也要求聚合物分子量可控制,以及具有良好的形态,以使工艺稳定,提高运转效能。这是未来超高分子量聚乙烯催化剂发展的方向,要求催化剂需具有较高的机械磨损强度并具有良好的颗粒形态。
技术实现要素:
12.发明所要解决的问题
13.在此,近年来,对于使用了超高分子量聚乙烯的、例如纤维、二次电池用隔膜、压缩成型品、柱塞挤出品等成型体而言,对在提高其性能的同时进一步提高生产率的要求正在提高,但目前已有的聚烯烃催化剂催化活性较低,并且所制备的超高分子量聚乙烯在颗粒形态、分子量级别和粘度等性能方面不能达到下游应用的需求。
14.本发明是鉴于上述问題完成的,其目的在于,提供一种活性氯化镁载体和相应的催化剂组分,以及用其制备得到的烯烃聚合催化剂、乙烯聚合催化剂,同时提供一种超高分子量乙烯类聚合物以及使用了超高分子量乙烯类聚合物的成型体,所述超高分子量乙烯类聚合物粉末能够提高使用了超高分子量聚乙烯的、例如二次电池用隔膜、纤维、压缩成型品、柱塞挤出品、纤维编织品和纺织品等成型体的性能并且提高其生产率。
15.于是,本发明人等为了实现所述课题,反复进行了深入研究,发现了一种原位合成的活性氯化镁,能够用于制备烯烃聚合的催化剂,并且所制备的聚乙烯聚合物,具有400,000-1300,000的粘均分子量,堆密度可达0.45g/cm3以上,颗粒形态也得到了很大程度的改善,粒径控制在120μm以下。
16.发明人惊奇地发现通过使用上述的特定超高分子量乙烯类聚合物粉末,能够解决目前应用中所存在的问题。具体而言,发现了:通过使用特定超高分子量乙烯类聚合物粉
末,缩短了其成型体、特别是二次电池用隔膜、纤维、压缩成型品、柱塞挤出品的制造时的冷却工序时间,并且在湿式挤出时在以往的超高分子量乙烯类聚合物粉末中容易发生溶胀不良,但本发明的超高分子量乙烯类聚合物粉末却呈良好的溶胀状态并且缩短了挤出时间。此外,发现了:例如对于二次电池用隔膜而言,使用公开的技术是困难的,但通过使用特定的超高分子量乙烯类聚合物粉末,能够减小二次电池用隔膜的平均孔径并且能够使之均匀,由此能够提高二次电池用隔膜的性能。
17.本发明人惊奇地发现,用原位合成的活性氯化镁可以作为催化剂载体。这种活性氯化镁不同于普通无水氯化镁,具有γ、β型或δ型结构,或混合构型,并具有较大的比表面积。采用这种载体制备出的镁醇合液再与四氯化钛作用,在酸酐晶体析出剂的协同作用下,可制备出形态良好的催化剂。晶体析出剂的作用是载体析出结晶化的有序析出,这样制备出的催化剂晶体性强,比表面积大,颗粒结实。采用这种方式所制备的催化剂可以解决制备超高分子量聚乙烯纤维料、隔膜料存在的诸多问题。这种含有新型载体的催化剂,具有控制催化剂动力学和聚合物分子量的催化剂前体,该催化剂前体具有颗粒结实、催化活性高、动力学曲线平稳可控等优点,使用其制得超高分子量聚乙烯的堆密度高、粒度分布均匀细小、分子量高且可控。
18.本发明的目的是提供一种催化剂载体,该载体包括原位活性氯化镁成分,所述的原位活性氯化镁的组成为:
19.(mgcl2)(r1mgcl)
a
mg
b
[ti(or2)4)]
c
[si(or3)4]
d
,
[0020]
其中r1、r2、r3可相同可不同,为c
1-12
烷基,a为0.02-1,b为0-0.5,c为0-0.8,d为0-0.8。
[0021]
优选地,所述原位活性氯化镁为(mgcl2)(bumgcl)
0.59
或(mgcl2)(bumgcl)
0.58
mg
0.08
[ti(oc4h9)4)]
0.07
[si(oc2h5)4]
0.23
,其中bu即为正丁基的缩写。
[0022]
所述原位活性氯化镁的比表面积为85-110m2/g。
[0023]
优选地,所述原位活性氯化镁的孔体积为50-70ml/g;优选地,所述原位活性氯化镁的粒径为2-8μm。
[0024]
本发明的原位活性氯化镁采用如下制备方法制得,镁粉先经单质碘活化,再与氯代烷烃反应制得,或在加入单质碘进行所述活化的同时还加入钛酸酯和硅酸酯反应制得;优选反应条件为氮气保护及无水条件下进行。
[0025]
本发明的另一个目的在于提供一种催化剂前体,该催化剂前体包括前述的原位活性氯化镁载体和酸酐。
[0026]
优选地,所述酸酐选自降冰片烯酸酐、邻苯二甲酸酐、马来酸酐及其混合物;优选地,基于每摩尔活性氯化镁,所述酸酐衍生物的用量为0.05-1.0摩尔。
[0027]
优选地,所述的催化剂前体进一步包括有机醇类化合物、给电子体有机硅化合物、钛化合物。
[0028]
优选的,所述有机醇类化合物为roh,其中r为c
2-c
16
烷基;例如所述有机醇为乙醇、丙醇、丁醇、己醇、2-甲基成醇、正庚醇、异辛醇或正辛醇,或其混合物。
[0029]
优选的,所述有机硅化合物为一种或多种选自具有分子式(r)
n
si的化合物的组的有机硅化物,其中,n为1-4的整数,r相同或不相同,独立的选自:任选被一个或多个rs取代的如下基团:氧、c
1-c
12
烷基、c
2-c
12
烯基、c
2-c
12
炔基、c
3-c
10
环烷基、c
6-c
12
芳基、c
1-c
12
烷氧
基、c
6-c
12
芳氧基;其中rs为卤素、c
1-c
12
烷基、c
1-c
12
烷氧基、c
3-c6环氧基c
1-c
12
烷氧基。
[0030]
例如,所述有机硅化合物选自:二甲基二甲氧基硅烷、二丙基二甲氧基硅烷、二异丙基二甲氧基硅烷、异丁基二甲氧基硅烷、二丁基二甲氧基硅烷、环已基甲基二甲氧基硅烷、环已基异丙基二甲氧基硅烷、环戊基异丁基二甲氧基硅烷、环戊基异丙基二甲氧基硅烷、环戊基丁基二甲氧基硅烷、环戊基丙基二甲氧基硅烷、二环戊基二甲氧基硅烷、二苯基二甲氧基硅烷、苯基三甲氧基硅烷、甲基三甲氧基硅烷、丁基三甲氧基硅烷、异丁基三甲氧基硅烷、γ-氯丙基三甲氧基硅烷、γ-(2,3环氧丙氧)丙基三氧基硅烷、二甲基二乙氧基硅烷、二丙基二乙氧基硅烷、二异丙基二乙氧基硅烷、异丁基二乙氧基硅烷、二丁基二乙氧基硅烷、环已基甲基二乙氧基硅烷、环己基异丙基二乙氧基硅烷、环戊基异丁基二乙氧基硅烷、环戊基异丙基二乙氧基硅烷、环戊基丁基二乙氧基硅烷、环戊基丙基二乙氧基硅烷、二环戊基二乙氧基烷、二苯基二乙氧基硅烷、苯基三乙氧基硅烷、甲基三乙氧基硅烷、丁基三乙基硅烷、异丁基三乙氧基硅烷、γ-氯丙基三乙氧基硅烷、乙烯基三乙氧基硅烷、乙烯基三甲氧基硅烷、四甲氧基硅烷、四乙氧基硅烷/及其混合物;
[0031]
优选的,所述钛化合物具有(ro)
m
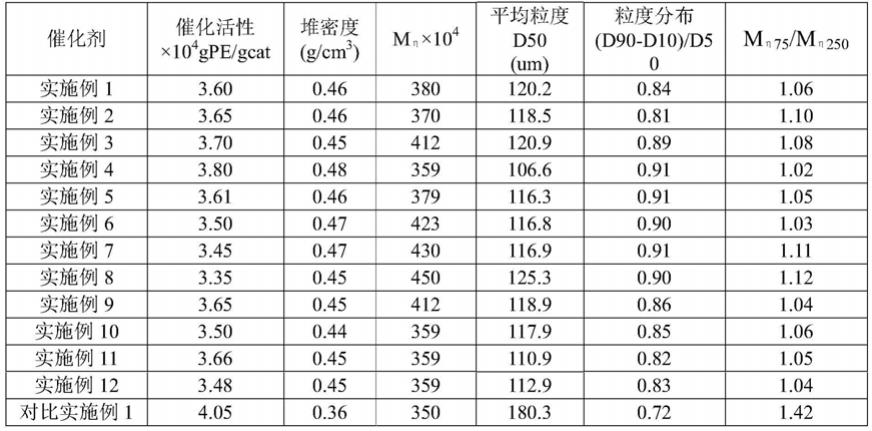
ticl
4-m
,其中,m为0-4的整数;例如选自四氯化钛、四溴化钛、四碘化钛、四丁氧基钛、四乙氧基钛、一氯三乙氧基钛、二氯二乙氧基钛、三氯一乙氧基钛、及其混合物。
[0032]
优选的,基于每摩尔活性氯化镁,所述有机硅化合物的用量为0.05-1.0摩尔,优选的,基于每摩尔活性氯化镁,所述钛化合物的用量为1.0-80.0摩尔、更优选20.0-50.0摩尔。
[0033]
本发明的又一种目的在于,提供一种催化剂组分,具体采用前述催化剂前体制备而成。
[0034]
具体的,所述催化剂组分的制备包括如下步骤:将原位活性氯化镁与有机醇类化合物、给电子体有机硅化合物、作为晶体析出剂的酸酐和可选的钛化合物接触反应,得到所述催化剂组分。
[0035]
更具体的,包括如下步骤:
[0036]
(1)在烃类溶剂中,将活性氯化镁与有机醇类化合物反应,得到镁醇合物反应液;
[0037]
(2)将步骤(1)制备的镁醇合物反应液与给电子体有机硅化合物和晶体析出剂酸酐进行反应;
[0038]
(3)将步骤(2)中得到的反应液与钛化合物混合,进行预载钛反应;
[0039]
(4)将步骤(3)得到的反应液与给电子体有机硅化合物混合,进行反应,制得催化剂组分。
[0040]
所述步骤(1)中,所述烃类溶剂为c
4-18
脂肪烃,优选c
6-12
脂肪烃;优选的,所述步骤(1)中,所述反应温度为50-180℃,反应时间优选0.5-5小时;其中镁/醇的摩尔比为1:0.5-6,优选为1:2-4;每摩尔镁化合物使用0.2-1.0升烃类溶剂。
[0041]
所述步骤(2)、(4)中所述的有机硅化合物可相同可不同,其为如权利要求6中所定义的有机硅化合物;优选的,所述步骤(2)中,所述反应温度为20-100℃。
[0042]
优选的,在上述步骤(3)中,所述反应的温度为-30-10℃、更优选-20-0℃、最优选为-10-0℃;反应时间优选0.5-5小时,0.5-3小时。
[0043]
优选的,在上述步骤(4)中,将步骤(3)得到的反应液升温到60-130℃,继续反应,例如反应1-6小时;例如在1-4小时内将反应温度升到60-130℃;之后再与有机硅化合物进
行反应;反应温度优选为60-130℃、优选至90-110℃,反应时间优选为1-4小时;所述有机硅化合物的用量为步骤(2)中有机硅化合物用量的1/10-1/2,优选为1/8-1/4;优选的,将步骤(4)中得到的反应液过滤,再用溶剂洗涤、干燥,得到固体催化剂。
[0044]
本发明的又一种目的在于,提供一种催化剂体系,具体包括:
[0045]
a)前述的催化剂组分;
[0046]
b)助催化剂,其为金属有机化合物,优选有机铝化合物r
3-n
a1x
n
,其中x为卤素,r为c1-c12烷基,n为0-2的整数;其中助催化剂中的铝与催化剂活性组分中的钛的摩尔比为10-800,优选50-200,更优选80-160。
[0047]
本实施方式中,催化剂的平均粒径优选为0.1μm以上且20μm以下,更优选为0.2μm以上且16μm以下,进一步优选为0.5m以上且10m以下。如果平均粒径为0.1μm以上,则具有可以防止所得到的乙烯聚合物粒子的飞散、附着等问题的倾向。另外,平均粒径为10μm以下时,具有可以防止乙烯聚合物粒子变得过大而在聚合体系内沉降、以及在乙烯聚合物的后处理工序中的管线的堵塞等问题的倾向。催化剂的粒径分布优选尽可能窄,可以利用筛、离心分离、旋风分离器而除去微粉粒子和粗粉粒。
[0048]
本发明还涉及一种用途,具体是将前述的原位活性氯化镁催化剂载体、前述的催化剂前体、由所述的催化剂前体制备的催化剂组分以及上述的催化剂体系用于制备聚烯烃的用途,优选用于制备聚乙烯的用途。
[0049]
本发明还涉及一种制备烯烃聚合物的方法,具体是烯烃聚合过程中采用了前述的原位活性氯化镁催化剂载体、前述的催化剂前体、由所述的催化剂前体制备的催化剂组分以及上述的催化剂体系。
[0050]
优选地,制备烯烃聚合物的方法,聚合反应温度30-90℃,优选40-80℃;反应压力为:0.1-1.0mpa,优选0.2-0.8mpa。
[0051]
进一步地,制备烯烃聚合物的方法,是以多段聚合工艺制备超高分子量聚乙烯,所述方法包括:
[0052]
以一个聚合工序使乙烯聚合为特性粘度15-50dl/g的超高分子量聚乙烯;以及,在另一个的聚合工序在少量的氢气存在下使乙烯聚合为特性粘度2-10dl/g的高分子量聚乙烯。
[0053]
优选地,第一段聚合反应的工艺条件为聚合温度60-80℃,聚合压力在0.4-0.8mpa,聚合时间1.5-4.0小时。第二段聚合反应的工艺条件为聚合温度75-90℃,聚合压力在0.6-1.2mpa聚合时间0.5-3.0小时;优选的,第一段聚合产物占总聚合产物的重量百分比为40-90%,第二段聚合产物占总聚合产物的重量百分比为10-60%;优选的,第二段聚合反应中氢气的比例为氢气在气相中比例0.1-30mol%,优选0.2-20mol%,最优选0.3-10mol%。
[0054]
本发明还涉及一种烯烃聚合物,具体是采用上述的制备烯烃聚合物的方法制得的烯烃聚合物;优选地,所述烯烃聚合物的堆密度在0.45g/cm3以上,优选地,所述烯烃聚合物粉末的粒径在120μm以下。
[0055]
优选的,所述的烯烃聚合物为颗粒状超高分子量聚乙烯(uhmwpe),其具有如下性能:
[0056]
至少4dl/g的特性粘度(η),
[0057]
50至200m之间的中值粒径d50,
[0058]
低于10ppm的残余ti-含量,和
[0059]
低于1000ppm的总灰分含量;
[0060]
优选的,所述颗粒状uhmwpe具有至少400kg/m3的表观体积密度。
[0061]
本发明还涉及一种成型体,由上述的超高分子量聚乙烯材料制得;优选地,所述成型体为纤维、带或膜状。
[0062]
本发明还涉及上述成型体的制品,优选地所述制品选自由绳索、缆线、网、织物和防护用具例如防弹制品组成的组。
[0063]
在实际的实施过程中,制备聚乙烯纤维的方法可以为本领域常规使用的方法,例如,其方法可以包括:将8重量份的所述超高分子量聚乙烯粉末、0.7重量份的所述抗氧剂和92重量份的溶剂油置于插有温度计和排气玻璃管的三颈烧瓶中,将此三颈烧瓶置于套式恒温器中升温,在90℃下停留5min,使三颈烧瓶中的超高分子量聚乙烯粉末的悬浊液变成絮状。在变成透明溶液时,测定温度,该温度为最佳溶胀温度。在最佳溶胀温度下停留40-60min,搅拌速度为10020r/min。然后升温到180℃,再停留1h,观察溶液溶解状态及成丝状态。其中,溶解状态通过观察溶液是否透明、有无杂质或气泡判断;成丝状态通过将溶液经螺杆挤出后观察其拉丝判断。在290℃,螺杆挤出机开始工作,20min后经过滤器、计量泵等纺丝组件,单孔喷丝头有超高分子量聚乙烯纺丝原液流出,刚流出时超高分子量聚乙烯纺丝原液有气泡及杂质,经过一段时间稳定,超高分子量聚乙烯原液无气泡、透明,在凝固浴中进行牵伸,后绕过辊筒落在盛丝桶中形成冻胶丝。然后将冻胶丝在小烧杯中环绕起来,在通风厨里按冻胶丝与甲苯质量比1:20的比例把二甲苯加入烧杯中,超声萃取6min。再次加入二甲苯进行二次萃取,接着把萃取丝张紧缠绕在干燥的纸筒上,萃取结束后将萃取丝放入通风厨中室温下干燥3h。最后对冻胶丝进行超倍拉伸,采用分级拉伸的方法,一级拉伸时的温度为80℃,拉伸倍数为15倍,二级拉伸时温度为100℃,拉伸倍数为2倍,三级拉伸温度为110℃,拉伸倍数为1.5倍,测试各级拉伸的力学性能。
[0064]
在制备方法中,所述溶剂油的具体实例可以包括但不限于:白油、矿物油、石蜡油、液体石蜡、白矿油和白色油,最优选为矿物油
[0065]
此外,聚烯烃催化剂针对聚合度一般是有适应性要求的,比如上述背景技术中提及的用于制备超高分子量聚乙烯的催化剂不适于制备普通聚乙烯,并且,适于制备普通聚乙烯的催化剂一般不适于制备超高分子量聚乙烯。这是因为对于要生产低分子量聚乙烯产品需要氢调敏感性好以及共聚性能优越的催化剂体系,而一般用于制备超高分子量聚乙烯催化剂体系的氢调敏感性和共聚性能都较弱。本发明的催化体系能够克服上述矛盾,不但可以用于制备普通分子量聚乙烯,同时也可以用于制备超高分子量聚乙烯。
具体实施方式
[0066]
下面通过实例详细说明本发明,应该清楚地理解,这里所描述的本发明形式仅仅是说明性的,不意味着限制本发明。本发明包括了在权利要求范围内的所有改进。
[0067]
载体制备实施例1
[0068]
制备本发明所述的载体,取0.2mol镁粉置于经过氮气置换过的500ml三口烧瓶中,加入100ml的癸烷、0.02g碘,和少量氯代正丁烷;加热至75℃搅拌活化2小时,滴加1mol干燥
的氯代正丁烷,可观察到明显的反应,继续反应3个小时,过滤,将所得固体用己烷洗涤后干燥,得到载体a,元素分析结果显示其载体a组成为:(mgcl2)(bumgcl)
0.59
。
[0069]
载体制备实施例2
[0070]
制备本发明所述的载体,取0.2mol镁粉置于经过氮气置换过的500ml三口烧瓶中,加入150ml己烷6mmol钛酸正丁酯、36mmol正硅酸乙酯及0.02g碘,加热至75℃搅拌活化2小时,滴加1mol干燥的氯代正丁烷,可观察到明显的反应,继续反应3个小时,过滤,将所得固体用己烷洗涤后干燥,得到载体b,元素分析结果显示载体b组成为:(mgcl2)(bumgcl)
0.58
mg
0.08
[ti(oc4h9)4)]
0.07
[si(oc2h5)4]
0.23
。
[0071]
载体制备实施例3
[0072]
按实施例2的方法制备载体,不同的是钛酸正丁酯的加入量为3mmol,正硅酸乙酯的加入量为18mmol,干燥后得到活性載体c,元素分析结果显示载体c组成式如下(mgcl2)(bumgcl)
0.58
mg
0.04
[ti(oc4h9)4)]
0.04
[si(oc2h5)4]
0.12
。
[0073]
载体制备实施例4
[0074]
按实施例3的方法制备载体,不同的是钛酸正丁酯的加入量为3mmol,干燥后得到活性载体d,元素分析结果显示载体d其组成式如下:
[0075]
(mgcl2)(bumgcl)
0.58
mg
0.06
[ti(oc4h9)4)]
0.04
[si(oc2h5)4]
0.24
。
[0076]
制备催化剂实施例1
[0077]
把0.05mol(以mg计)活性mgc12载体a,20ml癸烷和26ml异辛醇(0.167mol),加热至130℃反应60分钟,降温至65℃,于此温度下加入15mmolγ-氯丙基三甲氧基硅烷和15mmol降冰片烯酸酐、继续反应60分钟,冷却至室温后。在-10℃下用90分钟时间缓慢滴加于200ml tic14中,滴加完毕后保持温度0℃下60分钟,然后用120分钟时间缓慢升温至110℃,在此温度下加入5mmol相同硅烷给电子体后,继续反应120分钟,得到固体催化剂,停止搅拌后,会发现固体催化剂颗粒沉降速度很快。反应结束后热过滤出固体催化剂。用己烷洗涤,每次40ml,至滤液基本为无色,其中游离钛含量小于0.3mg/ml,干燥后得固体催化剂。
[0078]
制备催化剂实施例2
[0079]
把0.05mol(以mg计)活性mgc12载体a,20ml癸烷和26ml异辛醇(0.167mol),加热至130℃反应60分钟,降温至65℃,于此温度下加入15mmolγ-氯丙基三甲氧基硅烷和10mmol降冰片烯酸酐、继续反应60分钟,冷却至室温后。在-10℃下用90分钟时间缓慢滴加于200ml tic14中,滴加完毕后保持温度0℃下60分钟,然后用120分钟时间缓慢升温至110℃,在此温度下加入5mmol相同硅烷给电子体后,继续反应120分钟,得到固体催化剂,停止搅拌后,会发现固体催化剂颗粒沉降速度很快。反应结束后热过滤出固体催化剂。用己烷洗涤,每次40ml,至滤液基本为无色,其中游离钛含量小于0.3mg/ml,干燥后得固体催化剂。元素分析:ti:7.2%、mg:15.5%、cl:64.5%、si:1.4%。
[0080]
制备催化剂实施例3
[0081]
把0.05mol(以mg计)活性mgc12载体a,20ml癸烷和26ml异辛醇(0.167mol),加热至130℃反应60分钟,降温至65℃,于此温度下加入15mmolγ-氯丙基三乙氧基硅烷和10mmol降冰片烯酸酐、继续反应60分钟,冷却至室温后。在-10℃下用90分钟时间缓慢滴加于200ml tic14中,滴加完毕后保持温度0℃下60分钟,然后用120分钟时间缓慢升温至110℃,在此温度下加入5mmol相同硅烷给电子体后,继续反应120分钟,得到固体催化剂,停止搅拌后,会
发现固体催化剂颗粒沉降速度很快。反应结束后热过滤出固体催化剂。用己烷洗涤,每次40ml,至滤液基本为无色,其中游离钛含量小于0.3mg/ml,干燥后得固体催化剂。元素分析:ti:7.3%、mg:15.2%、cl:64.6%、si:1.4%。
[0082]
制备催化剂实施例4
[0083]
把0.05mol(以mg计)活性mgc12载体a,20ml癸烷和26ml异辛醇(0.167mol),加热至130℃反应60分钟,降温至65℃,于此温度下加入10.0mmolγ-氯丙基三甲氧基硅烷和5mmolγ-氯丙基三乙氧基硅烷,10mmol降冰片烯酸酐、继续反应60分钟,冷却至室温后。在-10℃下用90分钟时间缓慢滴加于200ml tic14中,滴加完毕后保持温度0℃下60分钟,然后用120分钟时间缓慢升温至110℃,在此温度下加入5mmol相同硅烷给电子体后,继续反应120分钟,得到固体催化剂,停止搅拌后,会发现固体催化剂颗粒沉降速度很快。反应结束后热过滤出固体催化剂。用己烷洗涤,每次40ml,至滤液基本为无色,其中游离钛含量小于0.3mg/ml,干燥后得固体催化剂。元素分析:ti:7.1%、mg:16.1%、cl:64.2%、si:1.5%。
[0084]
制备催化剂实施例5
[0085]
与制备催化剂实施例4相同,只是将降冰片烯酸酐改为5mmol。元素分析:ti:7.2%、mg:15.8%、cl:64.6%、si:1.4%。
[0086]
制备催化剂实施例6
[0087]
与制备催化剂实施例4相同,只是将活性mgc12载体改为载体b。元素分析:ti:6.8%、mg:16.8%、cl:63.6%、si:1.6%。
[0088]
制备催化剂实施例7
[0089]
与制备催化剂实施例4相同,只是将活性mgc12载体改为载体c。元素分析:ti:6.7%、mg:17.2%、cl:63.2%、si:1.7%。
[0090]
制备催化剂实施例8
[0091]
与制备催化剂实施例3相同,只是将活性mgc12载体改为载体d。元素分析:ti:6.8%、mg:16.6%、cl:63.7%、si:1.6%。
[0092]
制备催化剂实施例9
[0093]
与制备催化剂实施例4相同,只是将降冰片烯酸酐改为邻苯二甲酸酐。元素分析:ti:7.2%、mg:15.8%、cl:64.7%、si:1.5%。
[0094]
制备催化剂实施例10
[0095]
与制备催化剂实施例4相同,只是将活性氯化镁载体a改为活性氯化镁载体b。元素分析:ti:7.06%、mg:16.8%、cl:64.0%、si:1.55%。
[0096]
制备催化剂实施例11
[0097]
与制备催化剂实施例4相同,只是将活性氯化镁载体a改为活性氯化镁载体c。元素分析:ti:6.98%、mg:17.02%、cl:64.5%、si:1.58%。
[0098]
制备催化剂实施例12
[0099]
与制备催化剂实施例4相同,只是将活性氯化镁载体a改为活性氯化镁载体d。元素分析:ti:7.01%、mg:16.3%、cl:64.5%、si:1.52%。
[0100]
对比实施例1
[0101]
与制备催化剂实施例4相同,只是将活性氯化镁改为市售无水氯化镁。元素分析:ti:6.8%、mg:15.5%、cl:64.7%、si:1.5%。
[0102]
采用催化剂实施例1-12及对比实施例1的催化剂制备超高分子量聚乙烯实施例13
[0103]
在500升不锈钢高压釜中,经氮气置换后,依次加入脱水己烷300升,三乙基铝的己烷溶液(按aiti摩尔比为100),以及上述实施例制备的催化剂1.5g,搅拌速度900转/分,升温至60℃,再通入乙烯至釜压为0.6mpa(表压),在70℃,保持釜压为0.6mp下聚合反应2小时后降温至室温,出料干燥后得到uhmwpe聚乙烯产品。对每个实施例催化剂聚合的产品进行催化剂活性测定、堆密度测量、粘均分子量测量、测量平均粒径和粒径分布等,结果详细见表1。
[0104]
方法测定,表观密度用astm-d-1895方法测定。聚乙烯粒度是由激光粒子分析器(mastersizer x,malvern)测定,其中d10、d50和d90分布是指各百分率10、50和90下的粒子的大小。d50定义为平均粒子分布,粒度分布定义为(d90-d10)/d50。粘均分子量按照astm d4020-05的标准采用高温型乌氏粘度计法进行粘度测定,毛细管内径为0.53mm,并使用m
η
=5.37
×
104·
[η]
1.37
进行计算。
[0105]
表1
[0106][0107]
这里需要说明的是,m
η75
/m
η250
是指将聚乙烯粉末利用筛孔尺寸为75μm的筛网进行分级时在筛网上残留的粉末的粘均分子量(m
η75
)与利用筛孔尺寸为250μm的筛网进行分级时通过筛网的粉末的粘均分子量(m
η250
)之比。当比值为0.7以上,表明能够抑制大粒径粉末的粘均分子量变得过高。其结果是能够进一步提高大粒径粉末在溶剂中的溶解性,能够减少溶解残留所导致的拉伸时的缺陷。另一方面,通过比m
η75
/m
η250
为1.4以下,能够抑制小粒径粉末的粘均分子量变得过高。其结果是能够抑制由小粒径粉末彼此的聚集所引起的在溶剂中的溶解性的降低。能够减少溶解残留所导致的拉伸时的缺陷。
[0108]
制备超高分子量聚乙烯釜内合金实施例14
[0109]
在500升不锈钢高压釜中,经氮气置换后,依次加入脱水己烷300升,三乙基铝的己烷溶液(按al/ti摩尔比为100),以及制备实施例3所制备的催化剂1.5g,搅拌速度900转/分,升温至60℃,再通入乙烯至釜压为0.7mpa(表压),在t=65℃,保持釜压为0.7mp下聚合反应一定时间后,待乙烯吸收掉40kg时(得到产品产量pe1=40kg),停止第一段反应,采样测试聚合物粘均分子量,样品标记为pe1m。多余的气体不排放掉,把反应温度升高为85℃,然后再通入乙烯维持压力釜压在0.5mpa进行聚合反应,当乙烯吸收总量达到60kg时放料,
此时产品产量pe2约60-40=20kg,样品标记为pe1。
[0110]
这样pe1/pe2=66/34。聚合结果和物理机械性能分别见表2和3。
[0111]
制备超高分子量聚乙烯釜内合金实施例15
[0112]
在500升不锈钢高压釜中,经氮气置换后,依次加入脱水己烷300升,三乙基铝的己烷溶液(按al/ti摩尔比为100),以及制备实施例3所制备的催化剂1.5g,搅拌速度900转/分,升温至55℃,再通入乙烯至釜压为0.6mpa(表压),在t=60℃,保持釜压为0.7mp下聚合反应一定时间后,待乙烯吸收掉40kg时(得到产品产量pe1=40kg),停止第一段反应,采样测试聚合物粘均分子量,样品标记为pe1m。多余的气体不排放掉,把反应温度升高为85℃,然后再通入乙烯维持压力釜压在0.5mpa进行聚合反应,当乙烯吸收总量达到60kg时放料,此时产品产量pe2约60-40=20kg,样品标记为pe2。
[0113]
这样pe1/pe2=66/34。聚合结果见表2,超高分子量聚乙烯釜内合金压模板的物理机械性能分别见表3。
[0114]
表2
[0115][0116]
表3
[0117]
实施例m
η
×
104抗冲击力(n)拉伸强度(mpa)断裂伸长率%实施例4359138538.45310实施例14372142540.84330实施例15490149943.17330
[0118]
超高分子量聚乙烯的强度以及耐磨性是随着分子量增加而增加,但随着分子量的增加,加工性越来越困难。釜内合金可以很好地解决这个问题。对于超高分子量聚乙烯而言,分子量越高,拉伸强度越高,而拉伸伸长率越小。实施例14和实施例15的分子量比实施例4要高,但伸长率却大。说明聚乙烯合金可以在不降低性能的条件下,提高加工性。
[0119]
表4
[0120][0121]
由于纺丝设备属于试验规模,拉伸条件有限,纤维的纤度最低只能做到25dtex,但这已足够与普通聚乙烯纤维比较其强度和韧性,以验证超高分子量聚乙烯釜内合金纤度的
可编织、可纺等应用的潜力。
[0122]
从上述试验结果可以看出,超高分子量聚乙烯釜内合金纤度即使为25和30dtex情况下,其纤维断裂强度依然分别达到了27.61和28.6en/dtex,能够与普通聚乙烯纤维相当,釜内合金中的高分子量成分的在分子量高达640万以上,依然显示出良好的强度和可纺性,表明超高分子量聚乙烯釜内合金可以提高纺丝的加工性并提高纤维产品的强度和韧性,这将大大拓宽超高分子量聚乙烯釜内合金在柔性防护产品、高端耐用品领域中的应用范围。
[0123]
本领域的技术人员应当理解,上述实施方式仅仅是为了清楚地说明本公开,而并非是对本公开的范围进行限定。对于所属领域的技术人员而言,在上述公开的基础上还可以做出其它变化或变型,并且这些变化或变型仍处于本公开的范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。