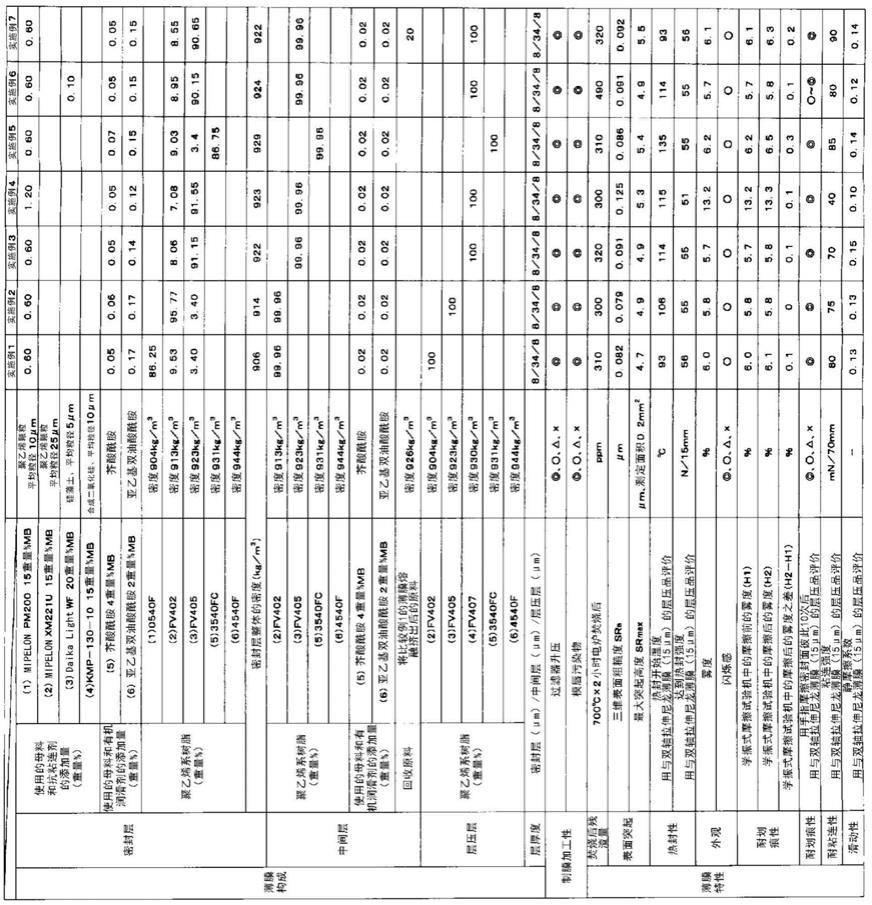
1.本发明属于分子生物学技术领域,具体涉及一种痕量单链核酸样本制备方法。
背景技术:
2.核酸是脱氧核糖核酸(dna)和核糖核酸(rna)的总称,是由许多核苷酸单体聚合成的生物大分子化合物,为生命的最基本物质之一。在天然存在的核酸中核糖核酸(rna)多以单链形式和发夹形式存在,也发现少量以环状形式存在。脱氧核糖核酸(dna)则以双螺旋的双链形式存在为主,也有部分以单链形式存在。在生物技术诸如核酸检测、体外诊断、基因测序等领域中,对核酸的各种处理过程中也会产生单链形式的核酸。比如,自然界中存在以单链dna或单链rna为遗传物质的生物如人类输血传染病毒(ttv)和微小病毒等等。对这些生物的遗传物质的测序研究使用传统的ngs测序前样本处理方法不容易实现。在法医领域,从腐烂的各种人源材料如骨骼、指甲、毛发等中提取出的dna或rna往往已经高度降解成了单链碎片化核酸,并且已经是痕量(如pg或者fg级别)。对这些痕量的碎片化核酸目前还没有一个高效的样本制备手段。 此外,在进行dna甲基化检测中也会涉及到将dna进行重盐转换的步骤,重盐转换后的dna绝大部分变成了比较短的碎片化单链dna。对这些单链核酸的处理目前也缺少一个特别高效的技术。
技术实现要素:
3.本发明的目的是提供一种可以对痕量核酸(dna或rna)进行样本制备的方法。该方法不依赖于dna的双链结构。该方法样本制备效率高,广泛适用于各种长度的痕量单链或双链dna及rna。
4.一种痕量单链核酸样本制备方法,包括以下步骤:(1)获取碎片化的,且经过5’端磷酸化和3’端羟基化处理的dna单链或者cdna;(2)步骤(1)得到的dna片段进行3’端辅助连接,延伸成dna双链;具体如下:体系中使用一种人工合成的核苷酸片段辅助子,以及dntp,且dntp是普通的dntp和热启动dntp的混合物,普通dntp选用四种dntp中的任一种、两种或者三种,热启动的dntp为选用的普通dntp对应剩余的三种、两种或者一种;在较低温度条件下利用所述的普通dntp进行dna片段的加尾反应;然后给体系一个较高的温度,使热启动的dntp进入到激活工作的状态;再给一个相对较低的温度,使辅助子与dna片段3’端加尾序列互补结合,最后由dna聚合酶分别以辅助子和dna片段为模板向前延伸,直至以辅助子为模板延伸出辅助子的互补链,并且以dna片段为模板延伸出dna片段的互补链,辅助子3’端含有与dna片段加尾互补的序列,且3’末端需要进行封闭,以防止体系中的加尾酶对辅助子进行3’加尾,延伸时解除3’末端的封闭;(3)步骤(2)得到的dna双链的5’端连接上接头,然后进行双模板底物扩增。
5.核酸碎片化:本发明的起始模板可以是双链或单链dna,也可以是rna。可以是各种长度为10bp
‑
1000bp。优选50bp
‑
800bp,进一步优选100bp
‑
300bp。如果是太长的dna或rna则
首先需要进行碎片化处理。碎片化处理的方案可以是基因检测行业内常用的方案。例如,长双链dna可以使用超声破碎的方法,内切酶酶切的方法等。长链rna可以使用二价金属离子加95℃以上高温处理的方法等。不仅限制于以上举例的处理方法,凡是能够对核酸进行有效碎片化处理的方法均可。如果核酸本身已经是较短的片段则不需要再进行碎片化处理。
6.cdna5’端磷酸化和3’端羟基化处理:对于rna分子,可以在进行逆转录时使用5’端磷酸化的逆转录引物,这样逆转录出来的产物是满足条件的5’端磷酸化和3’端羟基化的cdna。也可以在逆转录完成或者逆转录过程中同时使用t4 pnk酶等同样效果的酶进行5’端磷酸化修饰。对于dna分子,可以使用t4 pnk酶等同样效果的酶进行5’端磷酸化修饰和3’端羟基化处理。本步骤处理方式不仅限于所描述的方案,任何能达到同样效果的处理方案在本发明中都是可以执行的。
7.步骤(2)中:辅助子长度15nt
‑
120nt,优选36
‑
60nt;辅助子3’端至少第1个碱基(1个碱基效果最好,2
‑
3个碱基也可以)采用rna碱基进行封闭;所述的rna碱基为ra、rg、rc或ru。
8.步骤(2)中:dna片段3’端加尾序列长度范围1
‑
20个碱基,优选3
‑
10个碱基,辅助子3’端与dna片段3’端加尾序列互补结合的碱基序列在辅助子3’端rna碱基之后直到第14个碱基中的任意部分。
9.本发明所述的辅助子的3’端有部分碱基序列与dna的3’端的加尾序列互补。互补的意思是指,如体系中加入的普通dntp为datp,则辅助子的3’端对应的互补序列应为t碱基。该互补的碱基序列可以在辅助子3’端第2
‑
15碱基中的任意部分。例如可以是辅助子3’端第2
‑
8碱基、第2
‑
9碱基、第2
‑
10碱基、第2
‑
11碱基、第2
‑
12碱基、第2
‑
13碱基、第2
‑
14碱基、第2
‑
15碱基。辅助子3’端第1个碱基为rna碱基。该碱基可以是ra、rg、rc或ru,以防止体系中的加尾酶对辅助子或进行3’加尾。这是防止核酸样本形成接头二聚体的关键。
10.进一步地,通过限制普通dntp的量和/或增加辅助子的量来控制dna片段3’端加尾碱基数目,随着普通dntp的浓度降低,则dna片段3’端加尾碱基数目会减少;随着辅助子的浓度增加,则dna片段3’端加尾碱基数目会减少。
11.进一步地,体系中辅助子浓度为10nm
‑
20um之间,优选50nm
‑
10um之间,进一步优选100nm
‑
5um。
12.进一步地,反应体系中dna片段量1fmol
‑
1nmol/50ul。
13.上述的普通dntp选用四种dntp中的任一种、两种或者三种,热启动的dntp为选用的普通dntp对应剩余的三种、两种或者一种;例如:可以理解为:普通dntp选用一种,热启动dntp则选用另外的三种;普通dntp选用两种,热启动dntp则选用另外的两种;普通dntp选用三种,热启动dntp则选用另外的一种。
14.进一步地,体系中普通dntp的浓度为2um
ꢀ‑
2mm,优选10um
ꢀ‑
1mm之间,进一步优选30um
ꢀ‑
0.5mm之间,体系中普通dntp浓度是每一种热启动dntp单体的1倍
‑
3倍。
15.进一步地,步骤(2)中:首先在20℃
‑
37℃条件下进行dna片段的加尾反应5
‑
30分钟;然后在90℃
‑
95℃反应1
‑
5分钟,使热启动的dntp进入到激活工作的状态,同时使辅助子3’端的rna碱基从辅助子上掉下来,激活辅助子的延伸功能;再在45℃
‑
72℃反应2
‑
30分钟,使辅助子与dna片段3’端加尾序列互补结合,之后由dna聚合酶分别以辅助子和dna片段为模板向前延伸,直至以辅助子为模板延伸出辅助子的互补链,并且以dna片段为模板延伸出
dna片段的互补链。
16.进一步地,体系中对于dna片段加尾实现方式,包括:使用加尾酶,以体系中加入的普通datp、dttp、dgtp和dctp,4种dntp中的任一种、两种或三种为材料进行dna片段的3’端加尾;优选:体系中加尾酶(末端转移酶)的浓度为0.15u/ul
‑
15u/ul,优选0.4u/ul
‑
5u/ul,进一步优选0.6u/ul
‑
3u/ul。
17.进一步地,体系中用于聚合的聚合酶包括taq聚合酶中的至少一种,或者高保真聚合酶中的至少一种;taq聚合酶包括:2g robust dna聚合酶、rtaq dna聚合酶、taqb dna聚合酶中的一种或几种,高保真聚合酶包括:kapa hifi热启动高保真聚合酶(roche,#kk2602)、pfu dna聚合酶、phusion dna聚合酶中的一种或几种;优选taq dna聚合酶;优选:体系中聚合酶的浓度为0.01u/ul
‑
2u/ul;优选0.05u/ul
‑
1u/ul,进一步优选0.08u/ul
‑
0.5u/ul。
18.进一步地,如果步骤(2)延伸使用的是高保真聚合酶,延伸产物为平末端,则对应使用的连接接头是平末端接头;如果延伸使用的是taq聚合酶,延伸产物为3’有a碱基的粘性末端,则对应使用的连接接头是3’有t碱基的粘性末端接头。
19.本发明优选:延伸使用的是不具有3’到5’方向外切活性的聚合酶,例如taq dna聚合酶等,因为这一方案可以保证双链延伸产物之间以及双链接头之间不会自我连接,即可以保证只能是双链延伸产物和双链接头进行连接。
20.本发明优势:本发明体系中的dntp是普通的dntp和热启动dntp的混合物。例如:dntp是普通的datp和热启动的dttp、dctp、dgtp(如:trilink公司的热启动dntp)。在较低温度条件下适合dna加尾反应,合适的加尾酶只能将普通的datp加入到dna的3’端。然后给体系一个较高的温度,使热启动的dttp、dctp、dgtp进入到激活可以工作的状态。再给一个相对较低的温度,使辅助子与dna3’端加尾序列互补结合,之后由dna聚合酶沿着dna的3’端尾向前延伸,直至以辅助子为模板延伸出辅助子的互补链。该方案的好处是dna加尾、dna3’端辅助连接、双链延伸可以在一个反应体系中进行。提高了制备痕量核酸样本的效率,并且减少了纯化步骤。这对痕量的dna起始原料样本处理来讲意义重大。
21.该方案中的辅助子3’端需要进行封闭,以避免dna加尾反应把辅助子误认为是模板dna进行误加尾操作。辅助子的3’端最后的碱基为rna,在高温条件下(如95℃)使封闭的rna碱基掉下来,使辅助子露出3’端羟基,以供体系中聚合酶聚合,生成模板互补链。
22.本发明由于对核酸模板本身及其延伸互补链都连接了接头序列,因此可以使用通用引物进行扩增,最终在pcr扩增指数期内相比于只连接一条链(核酸模板本身或其互补链)的方案可以得到多一倍的扩增产量。
附图说明
23.图1是本发明以rna为例的技术流程图。
24.图2是本发明具体技术原理图。
25.图3是本发明实施例1的扩增产物胶图。
26.图4是本发明实施例2中体系s1的结果图。
27.图5是本发明实施例2中体系s2的结果图。
28.图6是本发明实施例2中体系s3的结果图。
29.图7是本发明实施例2中体系s4的结果图。
30.图8是本发明实施例2中体系s5的结果图。
31.图9是本发明实施例3中体系s1的结果图。
32.图10是本发明实施例3中体系s2的结果图。
33.图11是本发明实施例3中体系s3的结果图。
34.图12是本发明实施例4的扩增产物胶图。
35.图13是本发明实施例4的扩增产物测序结果图。
具体实施方式
36.以下结合实施例旨在进一步说明本发明,而非限制本发明。
37.实施例1长链rna分子样本制备使用商业化试剂盒抽提拟南芥mrna。取500pg mrna进行样本处理;逆转录引物p1序列:5’p
‑
nnnnnnnn
‑3’
。
38.1. 核酸碎片化反应体系如下:70℃反应1.5分钟;qiagen柱纯化。
39.2. rna逆转录反应体系如下:65℃反应5分钟,立即置于冰上;配如下反应试剂:
将上一步的12ul含rna的反应体系加入7.5ul试剂中;25℃反应2分钟后立即加入superscript iii reverse transcriptase 1ul。混匀后进行如下反应:25℃ 10分钟,42℃ 50分钟, 4℃保存;反应完成后qiagen柱纯化。
40.3.cdna3’端加尾和辅助生成辅助子a1序列:gtgactggagttcagacgtgtctcttccgatctttttrg,见seq id no.1,辅助子3’端为rna碱基g;反应体系如下:先将30ul cdna(浓度200pm)在pcr仪上95℃1分钟,4℃保存;将上述20ul反应混合液加入上述cdna中,混匀;pcr仪上37℃10分钟,95℃2分钟,72℃5分钟,4℃保存。
41.4. 5’端接头连接5’端接头adt1制作:
adt1
‑
u:aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct,见seq id no.2adt1
‑
d:p
‑
gatcggaagagcacacgtctgaactccagtcacatcacgatctcgtatgccgtcttctgcttg,见seq id no.3adt1
‑
u和adt1
‑
d等物质的量混合后在pcr仪上98℃2分钟,缓慢降低到室温使其退火成双链即可制成5’端接头adt1;配制如下反应体系:pcr仪上25℃15分钟,65℃20分钟,4℃保存;2x ampure xp beads纯化。
42.5.双模板底物扩增扩增引物序列如下:pcr
‑
f1:aatgatacggcgaccaccga,见seq id no.4pcr
‑
r1:caagcagaagacggcatacgagatcgtgatgtgactggagttcagacgt,见seq id no.5扩增反应体系如下:扩增程序如下:
最终扩增产物取5ul进行2%琼脂糖胶电泳。
43.实施例1实验结果见图3。
44.实施例2单核苷酸的浓度对3’端加尾碱基数量的影响取一段30nt的人工合成单链dna寡核苷酸。
45.依照加入单核苷酸浓度的不同做如下实验;反应体系如下:先将30ul(浓度10um)30nt的人工合成单链dna寡核苷酸在pcr仪上95℃1分钟,4℃保存;将上述20ul反应混合液加入上述dna中,混匀;pcr仪上37℃10分钟,95℃2分钟,72℃5分钟,4℃保存;将产物跑毛细管电泳,通过观察单链dna长度大小可以观察到不同的单核苷酸(datp)浓度对加尾碱基数量的影响。
46.实施例2实验结果见图4
‑
图8。
47.实施例2结果说明加尾碱基的数量与体系中单核苷酸的浓度有关系,浓度越高加尾的数量越多;而加尾数量的过多在ngs测序中会占据大量的数据量;这些数据属于无意义数据;因此,选择合适的单核苷酸的量,对于高效的反应是重要的。
48.实施例3辅助子的量对3’端加尾碱基数量的影响取一段30nt的人工合成单链dna寡核苷酸;依照加入辅助子的不同比例做如下实验;反应体系如下:先将30ul(浓度10um)30nt的人工合成单链dna寡核苷酸在pcr仪上95℃1分钟,4℃保存;将上述20ul反应混合液加入上述dna中,混匀;pcr仪上37℃10分钟,95℃2分钟,72℃5分钟,4℃保存;将产物跑毛细管电泳,通过观察单链dna长度大小可以观察到不同的辅助子和连接子的比例对加尾碱基数量的影响。
49.实施例3实验结果见图9
‑
图11。
50.实施例3结果说明加尾碱基的数量与辅助子在体系中的量有关系;辅助子在一定范围内浓度越高加尾的数量越少;因此,在本发明的实际应用中,选择合适的辅助子浓度对于高效的反应是重要的;这有助于提前终止3’端加尾反应。
51.实施例4循环血游离dna(cell
‑
free dna,即cfdna)甲基化样本处理使用zymo ez dna methylation
‑
gold kit甲基化试剂盒(zymo research,#
d5005)进行甲基化转换处理;甲基转换后的cfdna进行如下步骤操作。
52.1.dna3’端加尾和辅助生成辅助子a1序列:gtgactggagttcagacgtgtctcttccgatctttttrg,辅助子3’端为rna碱基g;反应体系如下:t4多聚核苷酸激酶可以使反应材料cfdna的5’端充分磷酸化;先将30ul dna(浓度3nm)在pcr仪上95℃1分钟,4℃保存;将上述20ul反应混合液加入上述cdna中,混匀;pcr仪上37℃10分钟,95℃2分钟,72℃5分钟,4℃保存。
53.2.5’端接头连接5’端接头adt1制作:adt1
‑
u:aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatctadt1
‑
d:p
‑
gatcggaagagcacacgtctgaactccagtcacatcacgatctcgtatgccgtcttctgcttgadt1
‑
u和adt1
‑
d等物质的量混合后在pcr仪上98℃2分钟,缓慢降低到室温使其退火成双链即可制成5’端接头adt1;配制如下反应体系:
pcr仪上25℃15分钟,65℃20分钟,4℃保存;2x ampure xp beads纯化。
54.3.双模板底物扩增扩增引物序列如下:pcr
‑
f1:aatgatacggcgaccaccgapcr
‑
r1:caagcagaagacggcatacgagatcgtgatgtgactggagttcagacgt扩增反应体系如下:扩增程序如下:最终扩增产物取5ul进行2%琼脂糖胶电泳,见图12。
55.4.将该cfdna甲基化处理后的扩增产物在illumina测序平台上进行wgbs(全基因组甲基化测序)双端150bp测序。数据唯一比对效率达到83.9%。见图13。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。