1.本发明涉及一种火麻仁中的木脂素酰胺类化合物及其制备方法和应用,主要是在治疗或预防神经退行性疾病的药物或保健食品中的应用,属于中医药制备技术领域。
背景技术:
2.火麻(cannabis sativa l.)又名大麻、胡麻、野麻、汉麻、线麻等,为桑科(moraceae)大麻亚科(subfam.cannabioideae)大麻属(cannabis)植物。火麻仁为火麻的干燥成熟果实,是火麻的习用药用部位,产于素有“长寿之乡”美称的广西巴马瑶族自治县,也是当地居民喜食的一种药食两用中药。火麻仁入药始载于《神农本草经》,列为上品,性甘、平,归脾、胃、大肠经,具有润肠通便的功效,用于血虚津亏、肠燥便秘等症的治疗。有研究表明,长期食用火麻仁对当地人的健康和长寿有一定的促进作用。据文献报道,火麻仁提取物中含有生物碱类、木脂素酰胺类、大麻素类、螺烷类、联苄类、黄酮类、甾体类及脂肪酸类等化学成分,现代药理学研究表明,火麻仁中的木脂素酰胺类化合物具有抗肿瘤、抗氧化、降糖、抗焦虑、神经保护等活性。然而火麻仁提取物成分复杂,并且现有的提取木脂素酰胺类化合物的技术繁琐复杂,提取回收率低,其方法还有待于改进,因此为了从火麻仁中发现新的具有神经保护活性成分,对火麻仁提取物进行了深入的研究。
技术实现要素:
3.为了克服现有技术的缺陷,本发明提供了一种火麻仁中的木脂素酰胺类化合物及其制备方法和应用,该方法以火麻仁(cannabis sativa l.)为研究对象,对其去油后的乙醇提取物的正丁醇萃取部位进行了分离和纯化,得到了六个新的木脂素酰胺类化合物,对这些化合物进行了结构鉴定和生物活性研究,发现部分化合物具有很好的神经保护活性,即有多种抗神经退行性疾病的活性,具有潜在的药用价值。
4.本发明通过如下技术方案实现:
5.本发明的一种火麻仁中的木脂素酰胺类化合物,其结构式为如下提供的1~6所示的化合物中的一种或几种的混合物;
6.[0007][0008]
本发明提供了一种火麻仁中的木脂素酰胺类化合物的制备方法,包括以下步骤:
[0009]
步骤1:将干燥的火麻仁粉碎,加入石油醚加热回流提取,得到去油后的火麻仁残渣和石油醚提取液;
[0010]
将去油后的火麻仁残渣加入乙醇水溶液进行再次加热回流提取,得到废渣和乙醇水提取液;
[0011]
将乙醇水提取液蒸馏去除溶剂,得到总浸膏;
[0012]
步骤2:将总浸膏加入水混悬,得到悬浊液;其中,总浸膏与水的固液比为(0.9~1.1)g:(9~11)ml;
[0013]
将悬浊液用水饱和的正丁醇进行萃取,萃取后,得到正丁醇层(cfe
‑
b);其中,按体积比,悬浊液:水饱和的正丁醇=(0.5~1.5):(0.5~1.5);
[0014]
将正丁醇层用液相色谱
‑
梯度洗脱法进行洗脱,得到9个流分,分别命名为cfe
‑
b1~cfe
‑
b9;
[0015]
步骤3:
[0016]
将cfe
‑
b3采用液相色谱
‑
梯度洗脱法再次进行洗脱分离,得到5个流分cfe
‑
b31~cfe
‑
b35;
[0017]
将cfe
‑
b4采用液相色谱
‑
梯度洗脱法再次进行洗脱分离,得到9个流分cfe
‑
b41~cfe
‑
b49;
[0018]
步骤4:
[0019]
(1)将cfe
‑
b33经过ods柱色谱分离,以甲醇
‑
水体系进行梯度洗脱,得到cfe
‑
b331~cfe
‑
b339;其中在甲醇
‑
水体系中,甲醇的体积百分比为10~100%;
[0020]
(2)将cfe
‑
b335经过sephadex lh
‑
20柱色谱分离纯化,以体积比为2:1~1:2的二氯甲烷
‑
甲醇体系进行等度洗脱,得到流分cfe
‑
b3351~cfe
‑
b3353;
[0021]
将cfe
‑
b337经过sephadex lh
‑
20柱色谱分离纯化,以体积比为2:1~1:2的二氯甲烷
‑
甲醇体系进行等度洗脱,得到流分cfe
‑
b3371~cfe
‑
b3377;
[0022]
(3)将cfe
‑
b3352经过高效液相hplc分离,得到化合物2;
[0023]
将cfe
‑
b3372经过高效液相hplc分离,得到化合物5和化合物3;
[0024]
将cfe
‑
b3373经过高效液相hplc分离,得到化合物4。
[0025]
所述的步骤1中,火麻仁与石油醚的固液比为(0.9~1.1)g:(9~11)ml;石油醚加热回流提取的加热温度为溶液沸点 (0~2)℃,回流提取的次数至少2次,每次回流提取的时间为1~4h;
[0026]
火麻仁残渣与乙醇水溶液的固液比为(0.9~1.1)g:(9~11)ml;其中,乙醇水溶液优选为乙醇体积百分比为70~80%的乙醇水溶液;再次加热回流提取的加热温度为溶液沸点 (0~2)℃,回流提取的次数至少2次,每次回流提取时间为1~4h。
[0027]
所述的步骤1中,所述的蒸馏优选为减压蒸馏。
[0028]
所述的步骤2中,对正丁醇层采用液相色谱
‑
梯度洗脱法进行洗脱分离,采用的为硅胶柱色谱柱,洗脱液为二氯甲烷
‑
甲醇溶液,梯度浓度变化为100:0,99:1,98:2,97:3,95:5,93:7,9:1,85:15,8:2,7:3,6:4,1:1,0:100。
[0029]
所述的步骤3中,对cfe
‑
b3再次进行洗脱分离,采用的为硅胶柱色谱柱,洗脱液为二氯甲烷
‑
丙酮溶液,梯度浓度根据cfe
‑
b3的极性,根据硅胶薄层色谱确定所需的洗脱浓度,更具体为93:7,9:1,85:15,8:2,7:3,6:4,1:1,0:100。
[0030]
所述的步骤3中,对cfe
‑
b4再次进行洗脱分离,采用的为硅胶柱色谱柱,洗脱液为二氯甲烷
‑
丙酮溶液,梯度浓度根据cfe
‑
b4的极性,根据硅胶薄层色谱确定所需的洗脱浓度,更具体为93:7,9:1,85:15,8:2,7:3,6:4,1:1,0:100。
[0031]
在步骤4的(3)中,对cfe
‑
b3352经过高效液相hplc分离,采用的流动相为乙腈的体积百分浓度为15%~20%的乙腈
‑
水溶液。
[0032]
在步骤4的(3)中,对cfe
‑
b3372经过高效液相hplc分离,采用的流动相为乙腈的体积百分浓度为23%~27%的乙腈
‑
水溶液。
[0033]
在步骤4的(3)中,对cfe
‑
b3373经过高效液相hplc分离,采用的流动相为乙腈的体积百分浓度为18%~22%的乙腈
‑
水溶液。
[0034]
进一步的,火麻仁中的木脂素酰胺类化合物的制备方法,还包括以下步骤:
[0035]
步骤5:
[0036]
(1)将cfe
‑
b43经过ods柱色谱分离,以甲醇
‑
水体系进行梯度洗脱,得到cfe
‑
b431~cfe
‑
b439;其中,甲醇
‑
水体系中,甲醇的体积百分比为10~100%;
[0037]
(2)将cfe
‑
b436经过凝胶柱色谱分离纯化,以体积比为2:1~1:2的二氯甲烷
‑
甲醇体系进行等度洗脱,得到流分cfe
‑
b4361~cfe
‑
b4365;
[0038]
(3)将cfe
‑
b4362经过高效液相色谱纯化,得到化合物1;
[0039]
步骤6:
[0040]
(1)将cfe
‑
b44经过ods柱色谱分离,以甲醇
‑
水体系进行梯度洗脱,得到cfe
‑
b441~cfe
‑
b439;其中,甲醇
‑
水体系中,甲醇的体积百分比为10~100%;
[0041]
(2)将cfe
‑
b443经过一次或两次高效液相色谱分离或纯化,得到化合物6。
[0042]
所述的步骤5的(2)中的,凝胶柱优选为sephadex lh
‑
20柱。
[0043]
在步骤5的(3)中,对cfe
‑
b4362经过高效液相hplc纯化,采用的流动相为乙腈的体积百分浓度为18%~22%的乙腈
‑
水溶液。
[0044]
在步骤6的(2)中,对cfe
‑
b443经过高效液相色谱纯化,采用的流动相为甲醇的体积百分浓度为38%~42%的甲醇
‑
水溶液或乙腈的体积百分浓度为18%~22%的乙腈
‑
水
溶液。
[0045]
本发明的一种药物组合物,包含上述的化合物1~6中的一种或几种。
[0046]
所述的药物组合物,以化合物1~6中的一种或几种作为活性成分,制备成口服液、胶囊、片剂、泡腾片、颗粒剂或各种已知剂型中的一种。
[0047]
所述的药物组合物,可以为医学药物、保健品、功能性食品中的一种。
[0048]
本发明的化合物3~5的火麻仁中的木脂素酰胺类化合物,以及以其作为活性成分的药物组合物的应用,用于制备治疗和/或预防神经退行性性疾病。
[0049]
所述的神经退行性性疾病,具体为阿尔茨海默症和/或帕金森病。
[0050]
本发明的火麻仁中的木脂素酰胺类化合物及其制备方法和应用,相比于现有技术,其有益效果为:所使用的试剂原料来源广泛,成本低,适宜放大生产,简单易行,操作简便,能够有效富集木脂素酰胺类化合物,且化合物纯度高,稳定性好。此方法在食品、保健品和医药等领域具有广阔的开发应用前景。
附图说明
[0051]
图1为本发明实施例中的火麻仁中的木脂素酰胺类化合物的制备方法的流程示意图;
[0052]
图2为化合物1~6对h2o2诱导pc12细胞损伤的保护作用;其中,(a)为化合物3、化合物5和化合物6对h2o2诱导pc12细胞损伤的保护作用,(b)为化合物2对h2o2诱导pc12细胞损伤的保护作用,(c)为化合物1和化合物4对h2o2诱导pc12细胞损伤的保护作用。
[0053]
图3为化合物1~6对mpp
诱导pc12细胞损伤的保护作用;其中,(a)为化合物3、化合物5和化合物6对mpp
诱导pc12细胞损伤的保护作用,(b)为化合物2对mpp
诱导pc12细胞损伤的保护作用,(c)为化合物1和化合物4对mpp
诱导pc12细胞损伤的保护作用。
具体实施方式
[0054]
下面结合实施例对本发明作进一步的详细说明。
[0055]
实施例1制备火麻仁的正丁醇提取物
[0056]
干燥的火麻仁(47.1kg)粗碎,加10倍量石油醚加热回流提取2次,每次2小时,得到去油后的火麻仁残渣(36.4kg)。火麻仁残渣加10倍量70%乙醇加热回流2次,每次2小时,合并提取液,减压回收至膏状,得到总浸膏(1.8kg)。浸膏用10倍量水混悬,用等体积的水饱和的正丁醇萃取3次,得到正丁醇层cfe
‑
b(690.7g)。
[0057]
实施例2
[0058]
一种火麻仁中的木脂素酰胺类化合物的制备方法,其流程示意图见图1,具体包括以下步骤:
[0059]
cfe
‑
b(690.7g)首先经过硅胶柱色谱分离,以二氯甲烷
‑
甲醇体系(二氯甲烷
‑
甲醇的体积比为100:0,99:1,98:2,97:3,95:5,93:7,9:1,85:15,8:2,7:3,6:4,1:1,0:100)依次进行梯度洗脱,每个体系洗4个保留体积,每个体积9000ml,共得到9个流分(cfe
‑
b1—cfe
‑
b9)。根据薄层层析色谱行为分析,在二氯甲烷
‑
甲醇体积比97:3处得到流分cfe
‑
b3(46.6g),在二氯甲烷
‑
甲醇体积比95:5处得到流分cfe
‑
b4(120.7g)。cfe
‑
b3(46.6g)和cfe
‑
b4(120.7g)进一步通过硅胶柱色谱分离,二氯甲烷
‑
丙酮梯度洗脱(二氯甲烷
‑
丙酮的体积
比为93:7,9:1,85:15,8:2,7:3,6:4,1:1,0:100),得到14个流分(cfe
‑
b31—cfe
‑
b35和cfe
‑
b41—cfe
‑
b49)。其中cfe
‑
b33为14.8g,cfe
‑
b43为1.7g,cfe
‑
b44为7.2g。
[0060]
cfe
‑
b33(14.8g)经过ods柱色谱分离,以甲醇
‑
水体系(甲醇
‑
水的体积百分含量为10%,20%,30%,40%,50%,60%,70%,100%)依次进行梯度洗脱,每个体系洗4个保留体积,每个体积400ml。根据薄层层析色谱显色行为分析,在体积百分含量为30%的甲醇
‑
水溶液中得到流分cfe
‑
b335,在体积百分含量为40%的甲醇
‑
水溶液中得到流分cfe
‑
b337。cfe
‑
b335(1.0g)进行进一步的sephadex lh
‑
20柱色谱分离纯化,二氯甲烷
‑
甲醇体系(体积比为1:1)进行等度洗脱,得到流分cfe
‑
b3352,cfe
‑
b3352(624.0mg)进行进一步的制备高效液相(hplc)的分离,以紫外检测器为检测手段,波长280nm,以体积百分含量为17%的乙腈
‑
水溶液为洗脱剂进行hplc制备,流速为4ml/min,收集26min的色谱峰,得到化合物2(67.5mg)。cfe
‑
b337(661.7g)进行进一步的sephadex lh
‑
20柱色谱分离纯化,二氯甲烷
‑
甲醇体系(体积比为1:1)进行等度洗脱,得到流分cfe
‑
b3372和cfe
‑
b3373,cfe
‑
b3372(214.6mg)进行进一步的制备高效液相(hplc)的分离,以紫外检测器为检测手段,波长210nm,以体积百分含量为25%的乙腈
‑
水溶液为洗脱剂进行hplc制备,流速为4ml/min,收集16min的色谱峰,得到化合物5(10.0mg),收集34min的色谱峰,得到化合物3(10.6mg)。cfe
‑
b3373(133.1mg)进行进一步的制备高效液相(hplc)的分离,以紫外检测器为检测手段,波长220nm,以体积百分含量为25%的乙腈
‑
水溶液为洗脱剂进行hplc制备,流速为4ml/min,收集20min的色谱峰,得到化合物4(20.0mg)。
[0061]
cfe
‑
b43(1.7g)经过ods柱色谱分离,以甲醇
‑
水体系(甲醇
‑
水的体积百分含量为10%,20%,30%,40%,50%,60%,70%,80%,100%)依次进行梯度洗脱,每个体系洗4个保留体积,每个体积100ml。根据薄层层析色谱显色行为分析,在体积百分含量为40%的甲醇
‑
水溶液中得到流分cfe
‑
b436。cfe
‑
b436(61.8mg)进行进一步的sephadex lh
‑
20柱色谱分离纯化,二氯甲烷
‑
甲醇体系(体积比为1:1)进行等度洗脱,得到流分cfe
‑
b4362,cfe
‑
b4362(27.9mg)进行进一步的制备高效液相(hplc)的分离,以紫外检测器为检测手段,波长280nm,以体积百分含量为20%的乙腈
‑
水溶液为洗脱剂进行hplc制备,流速为4ml/min,收集32min的色谱峰,得到化合物1(16.7mg)。
[0062]
cfe
‑
b44(7.2g)经过ods柱色谱分离,以甲醇
‑
水体系(甲醇
‑
水的体积百分含量为10%,20%,30%,40%,50%,60%,70%,80%,100%)依次进行梯度洗脱,每个体系洗4个保留体积,每个体积200ml。根据薄层层析色谱显色行为分析,在体积百分含量为40%的甲醇
‑
水溶液中得到流分cfe
‑
b443。cfe
‑
b443(1.5g)以紫外检测器为检测手段,波长280nm,以体积百分含量为40%的甲醇
‑
水溶液为洗脱剂进行hplc制备,流速为4ml/min,收集19min的色谱峰,得到cfe
‑
b4434,cfe
‑
b4434(242.5mg)进一步制备,以体积百分含量为20%的乙腈
‑
水溶液为洗脱剂进行hplc制备,流速为4ml/min,收集20min的色谱峰,得到化合物6(51.2mg)。
[0063]
实施例3化合物的结构鉴定
[0064]
化合物1和化合物2的氢谱(1h
‑
nmr)和碳谱(
13
c
‑
nmr)的核磁数据如表1所示。
[0065]
化合物3和化合物4的氢谱(1h
‑
nmr)和碳谱(
13
c
‑
nmr)的核磁数据如表2所示。
[0066]
化合物5和化合物6的氢谱(1h
‑
nmr)和碳谱(
13
c
‑
nmr)的核磁数据如表3所示。
[0067]
表1化合物1和化合物2的1h
‑
nmr和
13
c
‑
nmr的核磁数据(cd3od)
[0068][0069][0070]
表2化合物3和化合物4的1h
‑
nmr和
13
c
‑
nmr的核磁数据(cd3od)
[0071][0072]
[0073][0074]
表3化合物5和化合物6的1h
‑
nmr和
13
c
‑
nmr的核磁数据(cd3od)
[0075][0076]
本发明中所示化合物1~6的理化性质如下:
[0077]
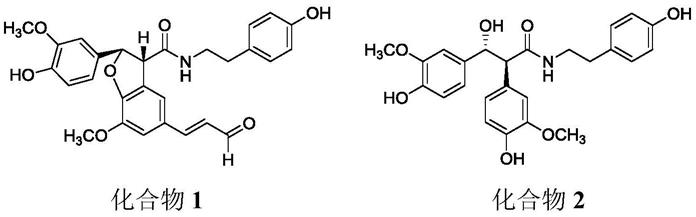
化合物1(cannabisin u)
[0078]
黄色粉末,hr
‑
esi
‑
ms(negative)给出m/z 488.1713[m
‑
h]
‑
(c
28
h
26
no7,计算值为488.1715),从而确定化合物的分子式为c
28
h
27
no7,计算其不饱和度为16。uv光谱给出λ
max
(logε)220(2.88),280(2.39)nm的木脂素酰胺类化合物特征吸收。
[0079]
化合物2(cannabisin v)
[0080]
淡黄色粉末,esi
‑
ms(positive)给出m/z 476.31[m na]
,提示化合物的分子量为453。hr
‑
esi
‑
ms(positive)给出m/z 476.1685[m na]
(c
25
h
27
no7na,计算值为476.1680),hr
‑
esi
‑
ms(negative)给出m/z 452.1709[m
‑
h]
‑
(c
25
h
26
no7,计算值为52.1715),从而确定化合物的分子式为c
25
h
27
no7,计算其不饱和度为
13。uv光谱给出λ
max
(logε)220(5.05),280(4.59)nm的木脂素酰胺类化合物特征吸收。
[0081]
化合物3(cannabisin p)
[0082]
淡黄色油状物,esi
‑
ms给出m/z 637.2[m na]
,613.0[m
‑
h]
‑
,提示化合物的分子量为614。hr
‑
esi
‑
ms给出m/z 613.2189[m
‑
h]
‑
(calcd 613.2186,c
34
h
33
n2o9),确定化合物的分子式为c
34
h
34
n2o9,计算其不饱和度为19。uv光谱给出λ
max
(logε)225(3.49),283(2.99)nm的木脂素酰胺类化合物特征吸收。
[0083]
化合物4(cannabisin x)
[0084]
白色粉末,esi
‑
ms(positive)给出m/z 619.1[m na]
,提示化合物的分子量为596。hr
‑
esi
‑
ms(negative)给出m/z 595.2084[m
‑
h]
‑
(c
34
h
31
n2o8,计算值为595.2075),从而确定化合物的分子式为c
34
h
32
n2o8,计算其不饱和度为20。uv光谱给出λ
max
(logε)220(4.03),280(3.82)nm的木脂素酰胺类化合物特征吸收。
[0085]
化合物5(cannabisin y)
[0086]
白色针晶(甲醇),esi
‑
ms(positive)给出m/z 306.2[m na]
,esi
‑
ms(negative)给出m/z 282.2[m
‑
h]
‑
,提示化合物的分子量为283。hr
‑
esi
‑
ms(positive)给出m/z 284.1281[m h]
(c
17
h
18
no3,计算值为284.1287),从而确定化合物的分子式为c
17
h
17
no3,计算其不饱和度为9。uv光谱给出λ
max
(logε)220(3.83),280(3.78)nm的木脂素酰胺类化合物特征吸收。
[0087]
化合物6(cannabisin z)
[0088]
黄色油状物,hr
‑
esi
‑
ms(positive)给出m/z 316.1185[m h]
(c
17
h
18
no5,计算值为316.1185),确定化合物的分子式为c
17
h
17
no5,计算其不饱和度为10。uv光谱给出λ
max
(logε)220(3.82),280(3.62)nm的木脂素酰胺类化合物特征吸收。
[0089]
实施例4化合物的神经保护活性
[0090]
(1)考察化合物对pc12细胞损伤程度,选取无损伤作用浓度进行后续实验,将pc12细胞接种在含10%胎牛血清(fetal calf serum,fbs)的rpmi 1640(roswell park memorial institute)培养基中,置于37℃恒温培养箱中培养12h,使用不同浓度的化合物(10μm、25μm、50μm、100μm)作用于pc12细胞,24h后检测pc12细胞存活率,据此选择进一步研究的化合物浓度。
[0091]
(2)考察化合物1~6对h2o2致pc12细胞损伤的保护作用
[0092]
采用h2o2诱导pc12细胞氧化应激,建立体外神经细胞损伤模型。将pc12细胞(1.0
×
104cells/ml,90μl/孔)接种于96孔板中恒温培养12h,加入250μm h2o2处理12h,随后选择不同浓度的化合物加入孔中,继续培养12h后检测细胞存活率。实验过程中的分组如下:(1)空白对照组:仅加入培养基;(2)阴性对照组:含h2o2(250μm)的培养基;(3)样品 h2o2处理组:不同浓度的样品 h2o2(250μm)。24h后,每孔加入mtt溶液,继续培养4h,吸弃上清液,每孔加入150μl dmso充分震荡待结晶完全溶解后,使用酶标仪在490nm波长下读取实验数据。
[0093]
细胞存活率(%)=(a
样品
–
a
空白对照
)/(a
阴性对照
–
a
空白对照
)
×
100%
[0094]
实验结果:采用mtt法考察化合物1~6进行神经保护活性测试(图2(a~c)),统计结果如表4所示。化合物3~5能够在一定程度上提高h2o2诱导pc12细胞损伤的生存率,提示
可能具有抗阿尔兹海默症的作用。
[0095]
表4化合物1~6对h2o2致pc12细胞损伤的保护作用
[0096][0097]
(3)单体化合物对mpp
诱导pc12细胞损伤的保护作用
[0098]
将pc12细胞接种在含10%胎牛血清(fetal calf serum,fbs)的rpmi 1640(roswell park memorial institute)培养基中,置于37℃恒温培养箱中培养。首先选择化合物的筛选浓度,继而采用mpp
诱导pc12细胞氧化应激,建立体外神经细胞损伤模型。将pc12细胞(1.0
×
104cells/ml,90μl/孔)接种于96孔板中恒温培养12h,随后加入500μm mpp
处理12h,继而选择不同浓度的化合物加入孔中,继续培养12h后检测细胞存活率。实验过程中的分组如下:(1)空白对照组:仅加入培养基;(2)阴性对照组:加入含mpp
(500μm)的培养基;(3)样品 mpp
处理组:不同浓度的样品 mpp
(500μm)。24h后,每孔加入mtt溶液,继续培养4h,吸弃上清液,每孔加入150μl dmso充分震荡待结晶完全溶解后,使用酶标仪在490nm波长下读取实验数据。
[0099]
细胞存活率(%)=(a
样品
–
a
空白对照
)/(a
阴性对照
–
a
空白对照
)
×
100%
[0100]
实验结果:采用mtt法考察化合物1~6对pc12细胞生存率的影响(图3(a~c)),统计结果如表5所示。化合物3和4能够在一定程度上提高mpp
诱导pc12细胞损伤的生存率,提示可能具有抗pd作用。
[0101]
表5化合物对mpp
诱导pc12细胞损伤的保护作用
[0102][0103]
表5数据表明化合物3和4能够在一定程度上提高mpp
诱导pc12细胞损伤的生存率,提示可能具有抗帕金森病的作用。
[0104]
对比例
[0105]
一种火麻仁中的木脂素酰胺类化合物的制备方法,同实施例1,不同之处在于,cfe
‑
b3(46.6g)进一步通过硅胶柱色谱分离,二氯甲烷
‑
丙酮梯度洗脱(二氯甲烷
‑
丙酮的体积比为95:5,9:1,85:15,8:2,7:3,6:4,1:1,0:100),不能得到洗脱产品。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。