1.本发明涉及复合光催化剂制备技术领域。具体地说是三氧化钼原位插层氮化碳复合催化剂及其制备方法。
背景技术:
2.从上世纪70年代至今,随着世界工业化的不断发展,人类面临着严重的能源匮乏问题,为解决这一难题,世界各地的研究学者开始寻找一种低污染、更耐久的新能源。经过不懈的努力,他们发现了零成本、零污染的太阳能。除此之外,由于现在工业化城市步伐的迅速加快,给环境带来巨大的问题,也给生物的生存条件加大了危机,限制了现代人类社会的可持续发展。
3.因为环境控制反应条件,中间化合物基本完全矿化,而且试验操作成本低,不会产生二次污染,所以光催化反应多被应用于降解有机污染物。光催化反应的本质实际上是氧化还原反应,我们知道,半导体材料的能带结构和导体的是不一样的,半导体的导带和价带间有一个禁带,光催化反应是在有光的条件下发生的,当光催化剂的带隙能量小于或等于光照射的能量时,就会产生空穴(h
)和光诱导电子(e
‑
)。导带上的电子是由价带产生并跃迁过来的,形成了光生电子
‑
空穴对。在内部电场的作用下,有一部分的光生电子和空穴将会复合,还有一部分会跃迁到催化剂表面发生氧化还原反应,从而降解表面的有机污染物。
4.石墨相氮化碳是一种聚合物半导体材料,具有良好的微观结构和化学功能,由于其对太阳光有响应,富含氮碳元素,不含金属元素,无毒等特征,可以作为一种潜在的光催化剂,可以有效的实现去污和能量转换,在环境治理和清洁能源方面有着潜在的重要作用。
5.虽然g
‑
c3n4聚合物非常容易制备、价格优惠、化学稳定性及热稳定性优良,而且光催化活性较好,但是它还是存在缺点:g
‑
c3n4聚合物的光生载流子和空穴的复合速率太快;由于比表面积较小,可见光响应范围有限,导致其光催化活性欠佳。因此,研究者们开发了一系列的方法对g
‑
c3n4进行改性,来改善其光吸收特性、比表面积和光生电荷分离效率,进而提高光催化效果。主要的改性方式有元素掺杂、形貌调控、异质结构造、z型体系构建、表面贵金属沉积和助催化剂等,但目前改性后的g
‑
c3n4聚合物大多存在制备方法复杂,光催化活性不理想或者稳定性差等缺陷。为此,申请人在安徽高校自然科学研究重点项目(项目编号:kj2019a0513)的资助下,对光复合催化剂及其制备方法进行了深入研究。
技术实现要素:
6.为此,本发明所要解决的技术问题在于提供一种对甲基橙和四环素等有机污染的降解速率快、光催化活性高的三氧化钼原位插层氮化碳复合催化剂及其制备方法,以解决当前氮化碳光催化活性及稳定性较差等问题。
7.为解决上述技术问题,本发明提供如下技术方案:
8.三氧化钼原位插层氮化碳复合催化剂的制备方法,包括如下步骤:
9.步骤a:向三聚氰胺中加蒸馏水,并加热搅拌,使三聚氰胺完全分散到蒸馏水中,得
到溶液a;
10.步骤b:向钼酸铵中加入蒸馏水,搅拌使钼酸铵完全溶解,得到溶液b;
11.步骤c:将溶液b缓慢逐滴转移至溶液a中,得到乳白色的混合液;
12.步骤d:将混合液加热搅拌,然后将混合液置于旋转蒸发仪上旋转蒸发干燥,使水分完全挥发,得到白色粉末状的前驱体;
13.步骤e:将前驱体置于马弗炉中进行煅烧,煅烧结束后得到黄色粉末状的三氧化钼原位插层氮化碳复合催化剂。与“分别单独制备三氧化钼和氮化碳,然后再将三氧化钼和氮化碳简单混合后进行煅烧”制备复合催化剂的方法相比,本发明的制备方法能够使得三氧化钼和氮化碳在原位空间上进行复合,避免了直接简单混合后煅烧这种方法的复合不均匀的情况,制备得到的复合催化剂性能也较好;本发明的制备方法是通过先制备得到一种特定的前驱体,然后通过煅烧前驱体煅烧得到原位复合材料,这与直接将三氧化钼和氮化碳混合后煅烧的复合方法完全不同。
14.上述三氧化钼原位插层氮化碳复合催化剂的制备方法,在步骤d中,mo元素以moo3计算,前驱体中moo3的质量与三聚氰胺的质量之比为0.1~1.0%。
15.上述三氧化钼原位插层氮化碳复合催化剂的制备方法,在步骤a中,三聚氰胺与蒸馏水的质量之比为1:(35~40);加热搅拌的温度为70~90℃,加热搅拌的时间15~25min。通过协同调控三聚氰胺与水的质量比、搅拌时间和搅拌温度,保证了三聚氰胺在水中形成均匀的水溶液,既不会溶解不完全又不会因加热水分蒸发过多而导致出现析出现象。
16.上述三氧化钼原位插层氮化碳复合催化剂的制备方法,在步骤a中,三聚氰胺与蒸馏水的质量之比为1:37.5;加热搅拌的温度为80℃,加热搅拌的时间20min。
17.上述三氧化钼原位插层氮化碳复合催化剂的制备方法,在步骤b中,钼酸铵与蒸馏水的质量之比为1:(200~2000),钼酸铵的搅拌溶解温度为18~25℃。将钼酸铵溶液逐滴转移至三聚氰胺溶液中时,若需要制备某一特定moo3含量的前驱体时,钼酸铵溶液浓度过低则需要钼酸铵溶液的体积增大,这不仅造成逐滴转移时间过长、制备效率低,而且还会降低前驱体的产率;钼酸铵溶液的浓度也不能过高,否则会导致反应沉淀后钼酸铵剩余,不仅造成浪费还会影响前驱体的纯度。
18.上述三氧化钼原位插层氮化碳复合催化剂的制备方法,在步骤b中,钼酸铵与蒸馏水的质量之比为1:250,钼酸铵的溶解温度为20℃。
19.上述三氧化钼原位插层氮化碳复合催化剂的制备方法,在步骤e中,煅烧方法为:先以5℃/min的升温速率升温至450~550℃,并保持温度不变煅烧3h;然后以5℃/min的降温速率降至室温,煅烧结束。通过调控升温速率和降温速率以及煅烧温度,使制备得到的复合催化剂产率较高且性能较好。在试验中发现,若升温速率大于5℃/min,会显著降低产物的产率,甚至完全煅烧掉,若升温速率小于5℃/min则容易导致前驱体煅烧不完全,致使前驱体不能完全烧结成目标复合催化剂,使产率下降。
20.上述三氧化钼原位插层氮化碳复合催化剂的制备方法,在步骤d中,混合液加热搅拌时的温度为80℃,加热搅拌时间为3h;旋转蒸发仪的旋蒸温度为70℃,转速为200r/min。若加热搅拌温度过低或者过高都会导致产物不纯净,若搅拌时间过短则反应不彻底。相比于过滤、洗涤再干燥的干燥方式,采用旋转蒸发的方式可以有效提高前驱体的产率;而当旋转蒸发的温度为70℃,转速为200r/min时,既保证了水分的快速去除,又不会影响前驱体的
产率和性能。
21.上述三氧化钼原位插层氮化碳复合催化剂的制备方法,在步骤a中,三聚氰胺与蒸馏水的质量之比为1:37.5,加热温度为80℃,搅拌时间20min;
22.在步骤b中,钼酸铵与蒸馏水的质量之比为1:250,钼酸铵的溶解温度为20℃;
23.在步骤d中,混合液加热搅拌时的温度为80℃,加热搅拌时间为3h;旋转蒸发仪的旋蒸温度为70℃,转速为200r/min;前驱体中moo3的质量与三聚氰胺的质量之比为0.8%;
24.在步骤e中,煅烧方法为:先以5℃/min的升温速率升温至480℃,并在480℃下煅烧3h;然后以5℃/min的降温速率降至室温,煅烧结束。
25.三氧化钼原位插层氮化碳复合催化剂,由上述三氧化钼原位插层氮化碳复合催化剂的制备方法制备得到。
26.本发明的技术原理:
27.三氧化钼是一种物理化学性质稳定、且能够吸收可见光的半导体材料,使用高温煅烧法,烧结一定量的钼酸铵就可以制备出具有光催化性能的moo3,这种方法非常简单而且合成的样品性能稳定。使用氮化碳与半导体moo3复合,利用二者能级位置的不同,经过焙烧复合后形成z型异质结构,这种结构能使光生载流子得到有效的分离,降低电子与空穴复合率,同时还能拓展光谱响应,从而增强对甲基橙溶液的光催化降解性能,这是因为该结构的moo3插层能有效地抑制光激发电子
‑
空穴对的复合,提高g
‑
c3n4的光吸收能力等;因此,将moo3插层到g
‑
c3n4的层间,可以显著提高光催化性能。
28.本发明的技术方案取得了如下有益的技术效果:
29.在安徽高校自然科学研究重点项目(项目编号:kj2019a0513)的资助下,本发明采用化学沉积法结合煅烧法制备得到了三氧化钼原位插层氮化碳复合催化剂,三氧化钼原位插层不仅提高了g
‑
c3n4的光吸收能力,而且有利于g
‑
c3n4的层间电荷分离,从而使该复合催化剂具有较高的光吸收能力和较高的电子空穴分离能力,对有机污染物表现出较好的光催化活性:对甲基橙溶液和四环素溶液均表现出了较好的降解效果,在300w汞灯照射1h时,mo
‑
cn
‑
0.8对甲基橙溶液和四环素溶液的降解率分别达到90%和93%;另外,本发明制备的复合催化剂在进行光催化时还具有较强的稳定性,可反复使用。
30.与“分别单独制备三氧化钼和氮化碳,然后再将三氧化钼和氮化碳简单混合后进行煅烧”制备复合催化剂的方法相比,本发明的制备方法能够使得三氧化钼和氮化碳在原位空间上进行复合,避免了直接简单混合后煅烧这种方法的复合不均匀的情况,制备得到的复合催化剂性能也较好;本发明的制备方法是通过先制备得到一种特定的前驱体,然后通过煅烧前驱体煅烧得到原位复合材料,这与直接将三氧化钼和氮化碳混合后煅烧的复合方法完全不同。
31.本发明在加热搅拌混合液使其进一步发生沉淀反应时,通过控制加热搅拌的温度和时间保证沉淀反应彻底进行,若加热搅拌温度过低或者过高都会导致产物不纯净,若搅拌时间过短则反应不彻底。本发明采用旋转蒸发的方式将沉淀反应得到的混合液中的水分挥发,这是因为技术人员在试验中发现,相比于过滤、洗涤再干燥的干燥方式,采用旋转蒸发的方式可以有效提高前驱体的产率;而当旋转蒸发的温度为70℃,转速为200r/min时,既保证了水分的快速去除,又不会影响前驱体的产率和性能。
32.本发明通过协同调控三聚氰胺与水的质量比、搅拌时间和搅拌温度,保证了三聚
氰胺在水中形成均匀的水溶液,既不会溶解不完全又不会因加热水分蒸发过多而导致出现析出现象;在配制钼酸铵溶液时,本发明控制钼酸铵与蒸馏水的质量之比为1:(200~2000),这是因为在将钼酸铵溶液逐滴转移至三聚氰胺溶液中时,若需要制备某一特定moo3含量的前驱体时,钼酸铵溶液浓度过低则需要钼酸铵溶液的体积增大,这不仅造成逐滴转移时间过长、制备效率低,而且还会降低前驱体的产率;钼酸铵溶液的浓度也不能过高,否则会导致反应沉淀后钼酸铵剩余,不仅造成浪费还会影响前驱体的纯度。
33.在对前驱体煅烧时,本发明通过调控升温速率和降温速率以及煅烧温度,使制备得到的复合催化剂产率较高且性能较好。在试验中发现,若升温速率大于5℃/min,会显著降低产物的产率,甚至完全煅烧掉,若升温速率小于5℃/min则容易导致前驱体煅烧不完全,致使前驱体不能完全烧结成目标复合催化剂,使产率下降。
附图说明
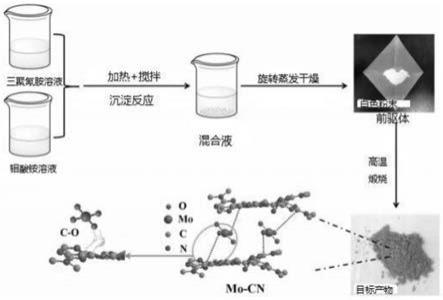
34.图1本发明复合催化剂的制备流程图及moo3插层g
‑
c3n4结构示意图;
35.图2本发明制备的mo
‑
cn
‑
0.8的前驱体的扫描电子显微镜图(10μm);
36.图3本发明制备的氮化碳的扫描电子显微镜图(1μm);
37.图4本发明制备的复合催化剂mo
‑
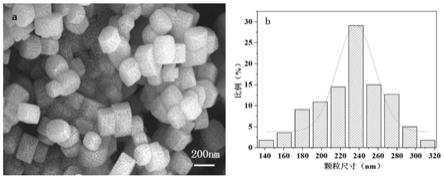
cn
‑
0.8的扫描电子显微镜图(1μm);
38.图5本发明制备的复合催化剂mo
‑
cn
‑
0.8的扫描电子显微镜图(1μm);
39.图6本发明制备的前驱体的xrd图;
40.图7本发明制备的复合催化剂的xrd图;
41.图8本发明制备的不同钼含量的复合催化剂的荧光光谱图;
42.图9本发明制备的不同钼含量的复合催化剂的uv
‑
vis漫反射光谱;
43.图10本发明制备的复合催化剂mo
‑
cn
‑
0.8的tg光谱图;
44.图11本发明制备的不同钼含量的复合催化剂的红外光谱图;
45.图12本发明中mo
‑
cn
‑
0.8光催化降解甲基橙的时间曲线图;
46.图13本发明中mo
‑
cn
‑
0.8光催化降解四环素的时间曲线图;
47.图14本发明中不同催化剂对甲基橙的降解度
‑
时间曲线图;
48.图15本发明中不同催化剂对四环素的降解度
‑
时间曲线图;
49.图16本发明中不同催化剂降解甲基橙的动力学曲线图;
50.图17本发明中不同催化剂降解四环素的动力学曲线图;
51.图18本发明中不同催化剂催化下甲基橙降解速率常数柱状图;
52.图19本发明中不同催化剂催化下四环素降解速率常数柱状图;
53.图20本发明中mo
‑
cn
‑
0.8对甲基橙的降解度与实验轮数关系图。
具体实施方式
54.1.三氧化钼原位插层氮化碳复合催化剂的制备方法
55.三氧化钼原位插层氮化碳复合催化剂的制备方法,包括如下步骤:
56.步骤a:向三聚氰胺中加蒸馏水,并加热搅拌,使三聚氰胺完全分散到蒸馏水中,得到溶液a;三聚氰胺与蒸馏水的质量之比为1:37.5,加热温度为80℃,搅拌时间20min;
57.步骤b:向钼酸铵中加入蒸馏水,搅拌使钼酸铵完全溶解,得到溶液b;钼酸铵与蒸
馏水的质量之比为1:250,钼酸铵的溶解温度为20℃;
58.步骤c:将溶液b缓慢逐滴转移至溶液a中,得到乳白色的混合液;
59.步骤d:将混合液加热搅拌,然后将混合液置于旋转蒸发仪上旋蒸使水分完全挥发,得到白色粉末状的前驱体;混合液加热搅拌时的温度为80℃,加热搅拌时间为3h;旋转蒸发仪的旋蒸温度为70℃,转速为200r/min;前驱体中mo元素以moo3计算,moo3的质量与三聚氰胺的质量之比为0.8%;
60.步骤e:将前驱体置于马弗炉中进行煅烧,煅烧方法为:先以5℃/min的升温速率升温至480℃,并保持480℃的温度不变煅烧3h;然后以5℃/min的降温速率降至室温;煅烧结束后得到的黄色粉末即为三氧化钼原位插层氮化碳复合催化剂。
61.本实施例采用原位合成法,结合单源预沉淀法制备了moo3插层g
‑
c3n4光复合催化剂,并考察了moo3插层对g
‑
c3n4的微观结构和光催化性能的影响。先将钼酸铵和三聚氰胺在一定的条件下进行反应(只改变钼酸铵的含量,其他条件均相同),得到前驱体(mamoo4),再将所制备的前驱体煅烧得到目标产物,即含有钼插层的氮化碳;然后通过不同的方法对纯氮化碳和含钼的氮化碳进行表征和测试,例如:x射线衍射光谱(xrd)、红外光谱(ir)、荧光测试、uv
‑
vis漫反射(drs)等;再用甲基橙溶液和四环素溶液在可见光下对催化剂进行光催化性能测试,找到光催化效果最好的样品,比较纯氮化碳和掺杂钼的氮化碳光催化性能的优劣;具体试验过程如下所述。
62.2试验内容
63.2.1试验仪器与试剂
64.表1.1主要仪器
[0065][0066]
表1.2主要试剂
[0067][0068]
2.2试验步骤
[0069]
2.2.1复合催化剂合成方法:化学沉积法
[0070]
(1)准备工作:用蒸馏水清洗好所用到的玻璃仪器,例如烧杯、量筒、胶头滴管等,然后放到鼓风干燥箱中烘干待用。
[0071]
(2)制备复合催化剂:准确称取4.0g三聚氰胺溶于150ml蒸馏水中,在恒温磁力搅拌器下,设置80℃温度,搅拌20分钟后三聚氢氨溶解,得到溶液a,溶液a澄清透明;准确称取0.005g钼酸铵溶于10ml蒸馏水中,20℃加热搅拌,使其溶解,得到溶液b,溶液b无色透明。用滴管将溶液b缓慢逐滴转移至溶液a中,溶液反应生成白色沉淀,得到乳白色浑浊的混合液。继续将混合液在80℃温度下加热搅拌3h,然后用旋转蒸发仪,设置70℃水温、200r/min转速,快速旋干,得到白色粉末,即为前驱体(mamoo4)。准确称取2.0g的前驱体(mamoo4)于坩埚中,放入马弗炉中,设置程序,以5℃每分钟的速度升温至480℃,在该温度下烧3h,然后再以5℃每分钟的速度降温,当温度降至室温后取出,可以得到目标产物,为黄色粉末(如图1)。mo元素以moo3计算,计算得,此比例下得到的前驱体中moo3/三聚氰胺为0.1wt%。用同样的方法步骤,改变钼酸铵的含量,分别制备出另外三种moo3/三聚氰胺为0.5wt%,moo3/三聚氰胺为0.8wt%,moo3/三聚氰胺为1.0wt%的不同钼含量的前驱体(mamoo4)。将最后得到的含钼的氮化碳(mo
‑
g
‑
c3n4)分别命名为mo
‑
cn
‑
0.1、mo
‑
cn
‑
0.5、mo
‑
cn
‑
0.8、mo
‑
cn
‑
1.0。
[0072]
(3)制备纯氮化碳催化剂:准确称取4.0g三聚氰胺于150ml蒸馏水中,在磁力搅拌器80℃温度下搅拌3h,得到澄清透明溶液,将该溶液放在旋转蒸发仪中,设置70℃水温、200r/min转速,旋干得到白色粉末,再将白色粉末放入马弗炉中,同样的升温速度480℃煅烧3h,得到淡黄色粉末。
[0073]
2.2.2光催化反应试验
[0074]
(1)准备工作:有机染料(甲基橙)溶液的制备:称量10mg甲基橙置于1000ml容量瓶中,加入蒸馏水定容,将容量瓶放入超声波仪器中,待甲基橙全部溶解即制备得到甲基橙溶液,取出容量瓶置于阴凉避光处待用;相同的方法可制备得到四环素溶液。
[0075]
(2)光催化反应试验:分别准确称取50mg mo
‑
cn
‑
0.1、mo
‑
cn
‑
0.5、mo
‑
cn
‑
0.8、mo
‑
cn
‑
1.0于四支石英管中,分别量取50ml甲基橙溶液置于上述四支石英管中;另取两支石英管,一支加入50mg纯氮化碳和50ml甲基橙溶液,另一支只加入50ml甲基橙溶液;将6支石英管同时放于光反应仪器中,在一定搅拌条件下(用恒温磁力搅拌器,加入1cm的磁子,搅拌速度是300
‑
400转/分)先暗反应30min,达成吸附
‑
脱附平衡后分别从6支石英管中各取样4ml,以300w的汞灯为光源进行光照,同时用滤光片过滤掉波长小于400nm的紫外光,以汞灯发出
的可见光进行光照,间隔相同的时间段(本实施例中间隔时间为20min)后再次从石英管中各取样4ml,每支石英管共取样5次。用12000r/min的高速离心机将取出的样品离心,使光催化剂与溶液分离,并取出上层清液,然后借助紫外
‑
可见分光光度计测定上清液的最大吸收波长处(463nm)的吸光度a,根据光照前后甲基橙溶液的吸光度变化,由降解度公式c/c0(%)=(a
t
/a0)
×
100%,可计算。a0,a
t
分别为光照前、光照后不同时刻甲基橙溶液在最大吸收波长处的吸光度值。
[0076]
3试验结果与讨论
[0077]
3.1前驱体和催化剂的sem图分析
[0078]
从图2至图5的sem图中可以看出,mo
‑
cn
‑
0.8的前驱体(图2)呈现出块状和类似石头状结构,表面呈现十分光滑的形态,具有比较宽的粒径分布。图3是纯氮化碳,可以看出它呈现层状结构,表面光滑。图4和图5为氮化碳复合催化剂mo
‑
cn
‑
0.8的sem图,与图3对比可知,三氧化钼的引入,对氮化碳的微观结构有比较大的影响,氮化碳复合催化剂的表面则变得十分粗糙,不再光滑,上面很多小颗粒附着,moo3被包裹在层与层之间。
[0079]
3.2前驱体和催化剂的xrd
[0080]
每一种物质的晶格类型及其晶胞尺寸都是一定的,每个晶胞中的原子位置及原子排列所构成的晶面也是固定的,因此每种物质都有其特定的衍射图形,即同一物质在一束确定波长的单色x射线中会产生确定的衍射图案,能在计算机谱图上观察到一系列的衍射峰。采用x射线衍射能谱仪对两种不同比例下合成的复合材料的物相结构进行分析,在2θ为5~70
°
的范围内对样品进行叠扫,扫描速度为每分钟8
°
。
[0081]
图6和图7分别是前驱体和催化剂的x射线衍射图谱,为了便于比较,本实施例还提供了纯水合三聚氰胺(melamine)的xrd图谱。图6中,mo
‑
cn
‑
x(x为0.8或1.0)样品的xrd图与纯三聚氰胺样品的xrd图非常相似,这应该是因为这些样品中钼酸盐的含量太少了。图7是氮化碳和氮化碳复合催化剂的结果,对比标准卡片,确定氮化碳的xrd衍射峰是正确的,可以看出两种复合催化剂在27.4处的峰值明显减小,这是moo3的成功插层影响的氮化碳的峰值。在图中没有moo3的衍射峰,这可能是因为插层的moo3太少了。
[0082]
3.3mo
‑
g
‑
c3n4复合光催化剂的荧光(pl)
[0083]
图8为纯g
‑
c3n4和不同比例下的mo
‑
g
‑
c3n4复合催化剂的荧光光谱,设置波长扫描范围为325nm
‑
575nm,显示了样品在300nm光激发下的pl光谱。纯g
‑
c3n4呈现一个以450nm为中心的强发射带,这意味着纯g
‑
c3n4具有较大的电子
‑
空穴复合率。由图8可知,mo
‑
cn
‑
0.1、mo
‑
cn
‑
0.5、mo
‑
cn
‑
0.8、mo
‑
cn
‑
1.0的发射带依次减小,即随着moo3插入的含量增大,这种发射带逐渐减小。该结果表明,moo3插层能有效地抑制光激发电子
‑
空穴对的复合。图8中,复合催化剂的发射峰对应的波长是434nm,与氮化碳的发射峰对应的波长相比,复合催化剂发射峰发生了蓝移;这可能是加入了三氧化钼后,二者相互作用,改变了氮化碳的荧光性质。
[0084]
3.4uv
‑
vis漫反射吸收光谱(drs)
[0085]
图9展示了不同样品的紫外可见漫反射(uv
‑
visdrs)图谱,设定波长扫描范围为300nm~800nm,利用外推法对样品的uv
‑
vis漫反射吸收峰做切线。由图可知,纯氮化碳对应的吸收阈值为459nm,mo
‑
cn
‑
0.5对应的吸收阈值为466nm,mo
‑
cn
‑
0.8对应的吸收阈值为470nm,mo
‑
cn
‑
1.0对应的吸收阈值为476nm;该结果表明,moo3插层显著提高了光催化剂的可见光吸收强度,使催化剂能够吸收更多的可见光。除此之外,样品的吸收边随mo含量的增
加而逐渐发生红移,说明moo3插层对g
‑
c3n4的能带结构有影响。这一现象证实了moo3和g
‑
c3n4之间存在化学相互作用,而不是物理混合。由半导体光催化剂吸收边与带隙能的关系:eg=1240/λg(λg为半导体光吸收阈值,eg为带隙能),由计算可得g
‑
c3n4、mo
‑
cn
‑
x(x依此为0.5、0.8、1)的带能隙分别2.70ev、2.66ev、2.64ev、2.61ev,预测mo
‑
cn
‑
1.0光催化活性最好,moo3插层使光的吸收增强,有利于充分利用太阳光,产生更多的载流子。
[0086]
3.5催化剂的tg分析
[0087]
当催化剂加热到一定温度时,所测的催化剂的质量就会有产生变化,此时得到的热重曲线就不是一条直线了。通过研究热重曲线,可以知道被测样品急剧变化的温度范围及在多少度时产生的变化,也可以根据失重的质量,计算所剩物质的实际浓度,计算由两种物质合成的复合物中每种物质的比例。
[0088]
根据图10的热重分析,可以计算得到样品中moo3的实际含量。图中给出了样品mo
‑
cn
‑
0.8的tg曲线。由图可知在500~650℃范围内发现了急剧的失重,根据图10的热重分析,计算得到mo
‑
cn
‑
0.8中moo3的实际浓度为2.56wt%。
[0089]
3.6催化剂的红外光谱分析:
[0090]
图11为g
‑
c3n4和不同比例下的mo
‑
g
‑
c3n4复合催化剂样品的红外光谱图,取约200mg纯kbr用玛瑙研钵仔细研磨成白色粉末,取1~2mg试样加入到kbr中,并研磨至体系分散均匀状态,将研磨后的样品置于干燥箱中烘干,使用压片机将样品压成均匀透明薄片,通过傅里叶变换红外光谱仪测定样品的红外光谱图。以连续频率的红外光照射样品时,样品中的分子会有选择性的吸收能使分子中某些基团振动能级跃迁的辐射能量,这将引起分子偶极矩的变化,最终实现分子的振动和转动能级从基态跃迁到较高能量的激发态。因此可以用傅里叶红外吸收光谱法来确定化合物分子中存在的原子基团及其在分子结构中的相对位置。
[0091]
由图11可知,mo
‑
cn
‑
x样品具有与纯g
‑
c3n4几乎相同的ir特征,例如n
‑
h伸缩振动(2900
‑
3600cm
‑1)、c
‑
n杂环骨架振动(1200
‑
1800cm
‑1)和三嗪环振动(808cm
‑1)。显然,moo3的插层并没有改变g
‑
c3n4的主体结构。然而,在c
‑
n振动中可以看出一些变化。例如,g
‑
c3n4的1236cm
‑1峰值移到mo
‑
c3n4‑
0.5的1244cm
‑1,移到了mo
‑
c3n4‑
1的1245cm
‑1。这种转变是由于moo3对g
‑
c3n4的强烈作用所致。
[0092]
3.7g
‑
c3n4及mo
‑
g
‑
c3n4光催化反应活性测定
[0093]
采用甲基橙和四环素对合成的g
‑
c3n4及mo
‑
cn
‑
0.1、mo
‑
cn
‑
0.5、mo
‑
cn
‑
0.8、mo
‑
cn
‑
1.0四种样品进行光催化活性试验测定。试验中,使用300w汞灯作为光源,暗反应30min,光反应每20min取一次样,取五次。将试验结果以降解度c/c0(%)=(a
t
/a0)
×
100%公式所得结果为纵坐标,反应时间为横坐标作图。其中,c0是初始甲基橙和四环素的浓度,c是加催化剂进行光催化降解后甲基橙和四环素的浓度;a0是初始甲基橙和四环素的吸光度,a
t
是加样品进行光催化降解后甲基橙和四环素的吸光度。
[0094]
3.7.1g
‑
c3n4及mo
‑
g
‑
c3n4在甲基橙溶液和四环素溶液下的光催化试验
[0095]
(1)g
‑
c3n4光催化降解甲基橙溶液:取两支容量为50ml的石英管,编号为
①②
,在
①②
号石英管中放入相同规格的搅拌子。称取50mg的g
‑
c3n4置于标号为
①
的石英管中,并加入10mg/l的甲基橙溶液50ml,其中
②
号石英管中只加入10mg/l的甲基橙溶液50ml,不加g
‑
c3n4,用来做纯甲基橙溶液对比试验。将两支石英管同时放入光化学反应仪中,在不开灯条
件下暗反应30min,取上清液4ml进行离心(8000r/min,5min),暗反应结束后,开灯,使用300w汞灯做光源进行光催化试验,打开制冷循环水,每隔20、40、60、80和100min各取一次上清液,进行离心(2次),用紫外
‑
可见分光光度计测定甲基橙溶液的吸光度。
[0096]
(2)mo
‑
g
‑
c3n4光催化降解甲基橙:取四支容量为50ml的石英管,在四支石英管中分别加入四种钼含量不同的mo
‑
g
‑
c3n4,再加入10mg/l的甲基橙溶液50ml,将四支石英管同时放入光化学反应仪中,在不开灯条件下反应30min,取上清液4ml进行离心(8000r/min,5min),暗反应结束后,打开灯源,使用300w汞灯做光源进行光催化试验,打开制冷循环水,每隔20、40、60、80和100min取一次上清液,进行离心(2次),用紫外
‑
可见分光光度计测定甲基橙溶液的吸光度。
[0097]
(3)g
‑
c3n4及mo
‑
g
‑
c3n4光催化降解四环素:重复上面(1)和(2)的步骤,只将甲基橙溶液换成四环素溶液,分别在暗反应30min,光反应20、40、60、80和100min时取上清液4ml进行离心(8000r/min,5min),然后用紫外
‑
可见分光光度计测定四环素的吸光度。
[0098]
图12和图13分别是300w汞灯的可见光照射下mo
‑
cn
‑
0.8对甲基橙和四环素的光催化降解时间曲线图,从图12和图13中可以看出,甲基橙的特征吸收波长在463nm处,四环素的特征吸收波长在361nm处。由图12可知,甲基橙溶液的浓度随着光催化时间的增长而降低;光照20min后,甲基橙溶液的降解度达到了40%,光照40min后,甲基橙溶液的降解度达到了72.5%,光照60min后,甲基橙溶液的降解度达到84.2%,光照80min后,甲基橙溶液的降解度达到89.3%,光照100min后,甲基橙溶液的降解度达到了90.7%(降解度c/c0(%)=(at/a0)
×
100%)。同样的,随着光催化时间的增长,四环素溶液的浓度也逐渐降低了,100min时,四环素溶液的降解度达到了93.6%。由此可以推理,在可见光下光催化足够长的时间后,甲基橙溶液及四环素溶液或许可以降解完全。
[0099]
图14和图15分别为5种样品对甲基橙和四环素催化降解的降解度时间曲线;从图中曲线可以看出来,在没有光催化剂的情况下,光照射100min后,甲基橙溶液和四环素溶液的浓度几乎没有任何的变化(图中,pure指代没有添加光催化剂的甲基橙溶液或四环素溶液)。加入催化剂后,甲基橙溶液在纯氮化碳下经过光照100min降解度可以达到63.6%,而四环素溶液在纯氮化碳下经过光照100min降解度可以达到88.24%。在两种溶液中,5种样品催化效果由大到小的顺序为:mo
‑
cn
‑
0.8>mo
‑
cn
‑
1.0>mo
‑
cn
‑
0.5>mo
‑
cn
‑
0.1>g
‑
c3n4,说明了,在相同的时间内催化降解甲基橙溶液和四环素溶液,复合的氮化碳比纯氮化碳的催化效果好,催化效果更加明显。其中,mo
‑
cn
‑
0.8样品对两种溶液的光催化活性最佳,仅经过光照100min降解度就分别达到了90.7%和93.6%,远远超过了纯氮化碳在100min时候的降解度。
[0100]
另外,甲基橙溶液在复合催化剂的催化下其降解效率比纯氮化碳明显高很多,g
‑
c3n4在100min时的降解度只有63.6%,而mo
‑
cn
‑
0.8在100min时的降解度达到了90.7%;四环素溶液在复合催化剂的催化下其降解效率和纯氮化碳相比变化不大,g
‑
c3n4在100min时的降解度为88.24%,mo
‑
cn
‑
0.8在100min时的降解度为93.6%,只增加了5.36%。说明在甲基橙溶液中复合催化剂的催化效果更加明显,而催化剂对四环素溶液的催化作用更好。
[0101]
图16和图17分别所制备的复合催化剂降解甲基橙溶液和四环素溶液的动力学曲线图。为了研究甲基橙降解的反应动力学,用一级模型拟合试验数据:ln(c0/c)=kt b。式中,c0和c分别是时间0和t时刻甲基橙和四环素溶液的浓度,k是表观一级速率常数。如图可
知,光催化时间(t)与ln(c0/c)的关系曲线几乎是一条直线。
[0102]
根据图16和图17中的斜率,可得到所制备样品对甲基橙和四环素的降解速率常数(k)。图18和图19是纯氮化碳和四种不同含量钼插层的氮化碳催化剂的一级反应速率常数柱状图。由图18可知,降解甲基橙时,g
‑
c3n4的速率常数k为0.009min
‑1,mo
‑
cn
‑
0.1的速率常数k为0.011min
‑1,mo
‑
cn
‑
0.5的速率常数k为0.0235min
‑1,mo
‑
cn
‑
0.8的速率常数k为0.03min
‑1,mo
‑
cn
‑
1.0的速率常数k为0.0255min
‑1;从图19可知,降解四环素溶液时,g
‑
c3n4的速率常数k为0.01828min
‑1,mo
‑
cn
‑
0.1的速率常数k为0.0191min
‑1,mo
‑
cn
‑
0.5的速率常数k为0.0201min
‑1,mo
‑
cn
‑
0.8的速率常数k为0.02329min
‑1,mo
‑
cn
‑
1.0的速率常数k为0.02108min
‑1。其中,mo
‑
cn
‑
0.8的速率常数k是最大的。在降解甲基橙时,这个速率常数数值达到了g
‑
c3n4的速率常数的3.3倍,mo
‑
cn
‑
1.0的速率常数也达到了g
‑
c3n4的速率常数的2.8倍。而降解四环素溶液时,5种催化剂的速率常数k相差不大,即降解速率差不多。
[0103]
然而,moo3含量(此处为1.0wt%)的进一步增加导致光催化活性的下降(图16和图17),这可能是由于moo3分子掺杂到g
‑
c3n4晶体中,会在晶体中产生点缺陷,这些点缺陷具有化学活性,为反应提供了更多的位点。另一方面,它们会在带隙中引入与缺陷相关的子能级,从而捕获电子或空穴。通过这种方式,介导了光生(或空穴)的分离和转移。当缺陷浓度超过阈值(此处为0.8wt%)时,这些缺陷会转化为电子空穴对的复合中心,从而降低系统的光催化活性。
[0104]
3.7.2mo
‑
cn
‑
0.8催化活性稳定性测试试验
[0105]
(1)由上面的试验可知mo
‑
cn
‑
0.8催化剂对甲基橙的光催化效果最佳,将甲基橙作为目标降解物进行三轮光催化循环试验。取三支相同的石英管,在里面分别放入三个规格相同的磁子,各加入50mg mo
‑
cn
‑
0.8催化剂,然后再加入10mg/l的甲基橙溶液各50ml。将三支石英管同时放在光反应仪器中进行暗反应30min,然后用300w汞灯作为光源,光反应100min后,取4ml上清液离心(8000r/min,5min),并测出离心后上清液在甲基橙溶液的最大吸收波长(463nm)处的吸光度。
[0106]
(2)对三支石英管里面的溶液进行离心,将沉淀分离出来,放在烘箱里面烘干。取二支相同的石英管,再次进行上面的试验,试验步骤都相同,最后测出离心后上清液在甲基橙溶液的最大吸收波长处(463nm)的吸光度。
[0107]
(3)对二支石英管里面的溶液进行离心,将沉淀分离出来,放在烘箱里面烘干。取一支石英管,再次进行上面的试验,试验步骤都相同,最后测出离心后上清液在甲基橙溶液的最大吸收波长处(463nm)的吸光度。用公式(c0‑
c)/c0,将三次循环试验的降解率对循环的次数作图,得到图20。
[0108]
用甲基橙为目标降解物,将mo
‑
cn
‑
0.8催化剂做三轮光催化循环试验后,可知三次催化剂的降解率分别为90.0%、89.4%、87.8%,相差不大,说明了mo
‑
cn
‑
0.8催化剂具有良好的催化稳定性能。
[0109]
4结论
[0110]
本实施例通过简单的化学沉积法,再通过煅烧法,制备纯氮化碳和含三氧化钼掺杂的氮化碳。通过红外、扫描电子显微镜、x射线衍射分析样品的内部结构和表面形态,通过固体紫外和荧光表征样品的光吸收和光致发光特性等。试验结果表明,基于mo
‑
cn
‑
0.8复合催化剂较高的光吸收能力和高的电子空穴分离能力,其光催化性能最好,对甲基橙和四环
素溶液都具有最高的降解度,在300瓦汞灯下照射一个小时,降解率可达90%和93%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。