1.本发明属于有机光电技术领域,具体涉及到一种基于杂原子取代的苯并噻二唑单元的共轭聚合物及其制备方法。
背景技术:
2.有机太阳能电池因具有制备成本低、功能和结构易于调节、柔韧性及成膜性好、易于实现大面积柔性器件制造等优点而备受关注。
3.在材料结构方面,共轭聚合物中的重原子取代效应逐渐成为研究热点。通过“重”原子(第via族中s以下)的引入,并采用相对简单的合成路线,进而产生具有高分子量、在可见光近红外区有强吸收、窄的最高占据分子轨道(homo)
‑
最低未占据分子轨道(lumo)间隙(<1ev)的给体材料。在广泛的给体重复单元中,少数结构在重原子取代化学中应用广泛,如芴和环戊联噻吩(cpdt)是两个备受关注的给体结构家族。类似地,2,1,3
‑
苯并噻二唑由于其吸电子能力强、成本低、合成简单,是最受欢迎的受体构筑单元之一,其单体同样可用来研究重16组原子取代对共聚物性能的影响。同时我们采用还原氧化两步反应制备硒类似物,其与母体苯并噻二唑互为等价电子。
4.在合成方法方面,迄今为止的d
‑
a共聚物主要是通过钯催化的stille缩聚反应,该反应涉及的合成锡化共聚单体需要有毒的锡试剂,并会产生化学计量的有毒有机锡废物,其处理成本高的同时会引起严重的环境问题。考虑到聚合物太阳能电池的巨大潜力和快速发展,迫切需要开发一种新兴的,绿色环保的聚合方法来扩量合成这些高性能共聚物。直接芳基化聚合(darp)方法使其可再生和可持续合成成为可能。darp的关键步骤涉及钯催化的区域选择性芳基碳氢键功能化,事实上,该方法消除了传统过渡金属促进的交叉偶联反应中所需的对空气和湿度敏感、昂贵且有毒的有机金属试剂的需要。在典型的darp过程中,在钯催化剂和碱的存在下,芳基卤化物与(杂)芳族化合物的碳氢键反应,以辅助碳氢键活化步骤,提供相应的偶联产物。同时其反应副产物是良性的,从而提供了总体上更简洁和原子经济的合成序列,这使得darp极具吸引力。然而,与小分子的c
‑
h官能化反应中副产物通常易于分离相比,大多数darp缺陷(例如,β缺陷、支化、交联、均偶联)仍会嵌在聚合物结构中,这是其大规模生产的主要瓶颈。本发明通过对原料投量比、溶剂、催化剂和膦配体的多次优化筛选在一定程度上解决了darp法在聚合物合成方面的共性问题,同时促使聚合物在可见
‑
近红外区范围内具有宽而强的吸收,良好的化学稳定性以及较低的homo能级。
技术实现要素:
5.本发明着重于结构类似的苯并噻二唑单元给体材料的合成化学和光电性质进行比较,其中在重复单元中进行了重16原子取代。同时旨在无需使用对环境有害的有毒有机锡化合物,采用低负载量的催化剂和磷配体进行合成,pd2(dba)3(5mol%)和p(2
‑
meoph)3(10mol%),增加了其实用性。尽管沿着共聚物主链存在一些“不希望的”活性碳氢位点,但
仍以优异产率和高分子量合成了两种代表性的共聚物。
6.为实现上述目的,本发明提供如下技术方案:
7.第一方面,本发明涉及基于苯并噻二唑或苯并硒二唑的共轭聚合物,所述共轭聚合物的结构式如式(ⅰ)所示:
[0008][0009]
其中,x为s或se,r为c1
‑
c30支化烷基链,n≥1。
[0010]
第二方面,本发明涉及的第一种给体材料:基于苯并噻二唑共轭聚合物的制备方法,包括如下步骤:
[0011]
按摩尔量份数,以1份的单体m1为标准,将其和新戊酸(1.2份)、碳酸钾(3份)、催化剂三(二亚苄基丙酮)二钯(0.05份)以及磷配体三(2
‑
甲氧基苯基)膦(0.1份),溶于无水邻二甲苯有机溶剂中。整个体系在无水和无氧气氛下进行,加热反应至接近胶状后,经过索氏提取进行提纯,真空干燥获得深蓝色的基于苯并噻二唑的共轭聚合物。所述加热条件具体为:90℃下反应24小时至胶状;所述索氏提取涉及的有机溶剂依次为甲醇、石油醚、氯仿。所述的单体m1如下所示:
[0012][0013]
其中,r为c1
‑
c30支化烷基链。
[0014]
所述的单体m1制备步骤如下:
[0015]
a、按摩尔量份数,将1份起始原料a与1份噻吩锡化物b以及0.02份钯催化剂加到有机溶剂中,以80℃反应6小时;反应结束后,粗产物经甲醇超声清洗后得到中间体c,其结构式为:
[0016][0017]
所述的起始原料a具有如下结构:
[0018][0019]
所述噻吩锡化物b为2
‑
(三丁基锡)噻吩,具有如下结构:
[0020][0021]
b、按摩尔量份数,以1份中间体c为标准,将其与4份三苯基膦溶于有机溶剂氯苯中,以140℃反应17小时;反应结束后,经硅胶柱提纯干燥后得到关环中间体。按摩尔量份数,以1份关环中间体为标准,将其与2.5份溴化烃d以及0.53份碘化钾和3份碳酸钾加到有机溶剂n,n
‑
二甲基甲酰胺中,以80℃反应17小时;反应结束后,经硅胶柱提纯干燥后得到单体m1,其结构式为:
[0022][0023]
其中,r为c1
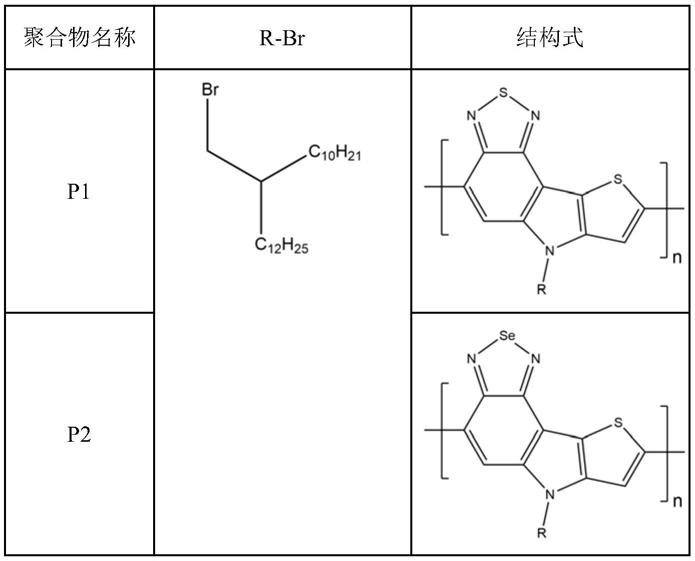
‑
c30支化烷基链。
[0024]
所述溴化烃d为c1
‑
c30溴化烷烃。
[0025]
所述步骤a有机溶剂为四氢呋喃,钯催化剂为双(三苯基膦)二氯化钯(ⅱ),起始原料a的合成参考专利文献:cn109879870a基于苯并噻二唑新型功能材料的合成及其应用。
[0026]
第二种给体材料:基于苯并硒二唑共轭聚合物的制备方法,包括以下步骤:
[0027]
按摩尔量份数,以1份的单体m2为标准,将其和新戊酸(1.2份)、碳酸钾(3份)、催化剂三(二亚苄基丙酮)二钯(0.05份)以及磷配体三(2
‑
甲氧基苯基)膦(0.1份),溶于无水邻二甲苯有机溶剂中。整个体系在无水和无氧气氛下进行,加热反应至接近胶状后,经过索氏提取进行提纯,真空干燥获得深蓝色的基于苯并硒二唑的共轭聚合物。所述加热条件具体为:90℃下反应44小时后升温至110℃反应24小时至胶状;所述索氏提取涉及的有机溶剂依次为甲醇、石油醚、氯仿。所述的单体m2如下所示:
[0028]
r为c1
‑
c30支化烷基链。
[0029]
所述的单体m2制备步骤如下:
[0030]
a、按摩尔量份数,以1份单体m1为标准,将其与40份锌粒加入到有机溶剂中,以80℃反应一定时间;反应结束后,由于双氨基在空气中不稳定,则粗产物通过快速减压过滤,乙酸乙酯和饱和食盐水多次洗涤,以及硅胶柱提纯干燥后得到中间体e,其结构式为:
[0031]
r为c1
‑
c30支化烷基链;
[0032]
b、按摩尔量份数,以1份双氨基单体e为标准,将其与6.7份二氧化锡以及三乙胺溶于有机溶剂中,回流反应24h;反应结束后,经硅胶柱提纯干燥后得到单体m2,其结构式为:
[0033]
r为c1
‑
c30支化烷基链。
[0034]
所述步骤a有机溶剂为乙酸,所述步骤b有机溶剂为三氯甲烷。
[0035]
本发明的显著优点:
[0036]
(1)本发明采用的原料均为普通化工原料,合成工艺简单且成熟,制备成本低;
[0037]
(2)本发明涉及的聚合物合成方法为直接芳基化聚合,该方法无需使用有毒锡试剂同时副产物均为良性,存在可持续、原子经济性高、环境友好等一系列独特优势;
[0038]
(3)本发明合成的聚合物主链具有共轭大π键的刚性平面结构,在分子骨架的一端进行成环进一步提高其平面性,同时增加柔性的烷基链使其在常见的有机溶剂中具有良好的溶解性,如:二氯甲烷、氯仿、氯苯等,有助于溶液加工处理,最后与重原子效应相结合,系统讨论光伏性质对给体结构中单个杂原子变化的依赖性;
[0039]
(4)本发明制备的聚合物具有宽而强的光吸收范围,良好的化学稳定性,较低的homo能级,适合应用于太阳能电池器件的给体材料。
附图说明
[0040]
图1为本发明实施例1中聚合物p1和p2的合成路线图。
[0041]
图2
‑
1、2
‑
2分别为本发明实施例1中单体m1和m2核磁共振氢谱。
[0042]
图3
‑
1、3
‑
2分别为本发明实施例1中聚合物p1和p2核磁共振氢谱。
[0043]
图4
‑
1、4
‑
2分别为本发明实施例1中单体m1和m2核磁共振碳谱。
[0044]
图5
‑
1、5
‑
2分别为本发明实施例1中聚合物p1和p2凝胶渗透色谱图。
[0045]
图6
‑
1、6
‑
2分别为本发明实施例1中聚合物p1和p2的热重曲线图。
[0046]
图7
‑
1、7
‑
2分别为本发明实施例1中单体m1和m2紫外吸收光谱。
[0047]
图8
‑
1、8
‑
2分别为本发明实施例2中聚合物p1和p2紫外吸收光谱。
[0048]
图9
‑
1、9
‑
2分别为本发明实施例2中聚合物p1和p2的循环伏安曲线图。
具体实施方式
[0049]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清晰、全面地描述。附图中给出了本发明较优的实施例,但本发明可以用不同的形式来实现,并不仅限于本文所描述的实施例。相反地,提供这些实施例的目的旨在使对本发明的公开内容的理解更加深入明晰。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改善和润色,这些也视为本发明的保护范围。
[0050]
在以下实施例中,努力确保所用数字(包括量、温度、反应时间等)的准确度,但应考虑一些不可抗力的实验误差和偏差。在以下实施例中所用的温度均以℃表示,压力为大气压或接近大气压。所用试剂均可从市场常规购得,所用试剂均为分析纯,所有反应均在惰性气氛下进行。
[0051]
实施例1、基于苯并噻二唑,苯并硒二唑单元的共轭聚合物的制备
[0052]
本实施例提供了两种基于苯并噻二唑或苯并硒二唑单元的可溶性共轭聚合物,其结构式如表1所示,其合成路线参见图1。
[0053]
表1
[0054][0055]
1.1、单体m1的制备
[0056]
两种聚合物的第一种单体m1的合成路线完全相同,其具体包括以下步骤:
[0057]
a、7
‑
溴
‑5‑
硝基
‑4‑
(噻吩
‑2‑
基)苯并[c][1,2,5]噻二唑的合成:
[0058]
在氩气条件下,将4,7
‑
二溴
‑5‑
硝基苯并[c][1,2,5]噻二唑(即起始原料a,合成参考专利文献:cn109879870a基于苯并噻二唑新型功能材料的合成及其应用)(3.3896g,10mmol),2
‑
(三丁基锡基)噻吩(3.7319g,10mmol)和50ml的干燥thf加入到干燥的schlenk反应瓶中。氩气鼓泡二十分钟除氧后加入催化剂双(三苯基膦)二氯化钯(ⅱ)(0.1404g,0.2mmol),80℃下加热搅拌6小时,通过tlc点板确认反应结束后冷却至室温。随后将反应混合物用甲醇洗一次,减压过滤后得到橙色固体,即7
‑
溴
‑5‑
硝基
‑4‑
(噻吩
‑2‑
基)苯并[c][1,2,5]噻二唑,产率为87.5%,结构式为:
[0059][0060]
b、4
‑
溴
‑6‑
(2
‑
癸基十四基)
‑
6h
‑
[1,2,5]噻二唑[3,4
‑
e]噻吩[3,2
‑
b]吲哚的合成:
[0061]
在氩气保护下,在干燥的schlenk反应瓶中,以30ml的干燥氯苯为溶剂,加入7
‑
溴
‑5‑
硝基
‑4‑
(噻吩
‑2‑
基)苯并[c][1,2,5]噻二唑(3.4219,10mmol)。用氩气鼓泡二十分钟除氧后加入三苯基膦(10.4916g,40mmol),将反应体系升温至140℃反应过夜。冷却至室温后,减压除去反应溶剂,残余物用硅胶柱层析分离提纯(石油醚:乙酸乙酯,v/v,4/1、2.8:1、2.3:1、二氯甲烷)得到粗产物。减压浓缩去除溶剂后加入甲醇超声多次,再用二氯甲烷冲洗直至滤液从橙色变为黄色为止,得到橙色固体,即关环中间体,这步产率为58.6%。50℃真
空干燥后,再将关环中间体(1.6112g,5.1942mmol)、2
‑
癸基十四烷基溴(5.4222g,12.9856mmol)和干燥dmf(320ml)加入到干燥的schlenk反应瓶中。用氩气脱气20min后再加入ki(0.457g,2.7529mmol)和k2co3(2.1537g,15.5827mmol)。所得溶液在80℃搅拌过夜。冷却至室温后,残留物用二氯甲烷和水萃取三次,稀盐酸和饱和食盐水各洗涤一次。收集到的有机层用无水硫酸镁干燥并减压过滤。残留物用硅胶柱色谱提纯,用二氯甲烷/石油醚(v/v,1/3、1/2、1/1.5)作为洗脱剂,除去溶剂后得到黄色蜡状,即4
‑
溴
‑6‑
(2
‑
癸基十四基)
‑
6h
‑
[1,2,5]噻二唑[3,4
‑
e]噻吩[3,2
‑
b]吲哚,产率为86.7%,结构式为:
[0062][0063]
1.2、单体m2的制备
[0064]
聚合物p2的单体m2的合成路线是在单体m1基础上,先进行还原开环,再氧化关环得到。其具体包括以下步骤:
[0065]
a、6
‑
溴
‑4‑
(2
‑
癸基十四烷基)
‑
4h
‑
噻吩并[3,2
‑
b]吲哚
‑
7,8
‑
二胺的合成:
[0066]
在氩气条件下,将4
‑
溴
‑6‑
(2
‑
癸基十四基)
‑
6h
‑
[1,2,5]噻二唑[3,4
‑
e]噻吩[3,2
‑
b]吲哚(0.5g,0.773mmol)和乙酸(45ml)加入到250ml双颈烧瓶中。用氩气液面下脱气20min后加入锌粒(2.0218g,30.9195mmol)。原料与乙酸的相容性较差,80℃加热反应一定时间(4
‑
48h),同时观察溶液体系颜色变化来初步判断反应程度:体系颜色从最初的无色透明,逐渐变为明黄色,最终变为乳白色,此时通过tlc监测确认反应结束。冷却至室温后快速过滤出锌粒和白色不溶物,即醋酸锌,得到的粗产物用乙酸乙酯和饱和食盐水多次洗涤,收集到的有机层用无水硫酸镁干燥并减压过滤,减压浓缩去除溶剂后得到黄色油状,即为6
‑
溴
‑4‑
(2
‑
癸基十四烷基)
‑
4h
‑
噻吩并[3,2
‑
b]吲哚
‑
7,8
‑
二胺,产率为99.6%,结构式为:
[0067]
无需进一步纯化,直接投入下一步。
[0068]
b、4
‑
溴
‑6‑
(2
‑
癸基十四烷基)
‑
6h
‑
[1,2,5]硒代己二唑并[3,4
‑
e]噻吩并[3,2
‑
b]吲哚的合成:
[0069]
将6
‑
溴
‑4‑
(2
‑
癸基十四烷基)
‑
4h
‑
噻吩并[3,2
‑
b]吲哚
‑
7,8
‑
二胺(0.4069g,0.6575mmol)、二氧化锡(99%,0.4888g,4.4056mmol)、三乙胺(1.4ml)和除好氧的无水氯仿(11ml)一次性加入到100ml的单口圆底烧瓶中,快速循环除氧多次后升温至回流反应24h。通过tlc监测确认反应结束,并将体系冷却至室温。减压过滤出白色不溶物,滤液用二氯甲烷和水萃取三次,无水硫酸镁干燥,减压过滤得到有机层。旋干溶剂后,粗产物用硅胶柱层析进行提纯,用石油醚/二氯甲烷(v/v,7/5)作为洗脱剂。减压浓缩除去溶剂,真空干燥得到橙色固体,即为4
‑
溴
‑6‑
(2
‑
癸基十四烷基)
‑
6h
‑
[1,2,5]硒代己二唑并[3,4
‑
e]噻吩并[3,2
‑
b]吲哚,产率为34.6%,结构式为:
[0070][0071]
1.3、聚合物p的合成
[0072]
对聚合物的聚合条件进行多次筛选和优化。首先是对催化剂的选择,采用多种钯催化剂进行试验,比如pd2(dba)3,pd(oac)2;其次对溶剂也尝试多次更换,比如甲苯、n
‑
甲基吡咯烷酮(nmp)、邻二甲苯;接着对磷配体也进行一定尝试,比如三(2
‑
甲氧基苯基)磷和三(邻甲基苯基)磷;最后对摩尔投料比也进行多次微调,最终优化后的聚合条件如下:
[0073]
在氮气保护下,分别将单体m1或m2(0.3092mmol)、新戊酸(0.3764mmol)、碳酸钾(0.9276mmol)以及1ml邻二甲苯投入到耐压管中,进行鼓泡除氧,最后加入催化剂三(二亚苄基丙酮)二钯(0.0141mmol)和磷配体三(2
‑
甲氧基苯基)膦(0.0309mmol),加热反应至接近胶状,将反应液冷却到室温,加入一定量甲醇沉淀,过滤出固体,依次用甲醇、石油醚、氯仿索氏提取,收集氯仿溶液,减压除去溶剂后再用甲醇沉淀得有金属光泽的深蓝色聚合物。p1和p2产率分别为99%和68%。
[0074]
原料为m1对应的加热条件具体为:90℃下反应24小时至胶状。
[0075]
原料为m2对应的加热条件具体为:90℃下反应44小时后升温至110℃反应24小时至胶状。
[0076]
实施例2、聚合物p1和p2的分子量
[0077]
采用凝胶渗透色谱仪来测试两种给体材料的分子量为:p1和p2的数均分子量(m
n
)分别为103.8kda、56.2kda,重均分子量(m
w
)分别为359.8kda、310.0kda,多分散指数pdi分别为3.46、5.51。
[0078]
实施例3、聚合物p1和p2的热重曲线
[0079]
从tga曲线可以看出,在惰性氛围下,聚合物p1和p2对应的5%质量损失分解温度分别达到435℃和350℃,表明具备良好的热稳定性,显示出二者满足作为给体材料应用于
有机太阳能电池器件中应用和其他光电器件的要求。
[0080]
实施例4、聚合物p1和p2的紫外
‑
可见吸收光谱
[0081]
分别在氯苯溶液和旋涂在载玻片上的固态薄膜状态下的紫外
‑
可见吸收光谱如图所示。在溶液中测量吸收特性,以确定重原子取代的效果。我们观察到,从s(695nm)移动到se(705nm)时,两种小分子给体的吸收最大值(λ
max
)移动到更长的波长。在聚合物吸收光谱中观察到从s到se的光谱吸收变化。由公式e
gopt
=hυ/λ
onsetfilm
=1240/λ
onsetfilm
可估算出二者的光学带隙(e
gopt
),与p2(1.46ev)相比,p1(1.57ev)的光学带隙能量(从吸收开始时测量)进一步说明了这一趋势。
[0082]
聚合物光谱比小分子光谱更复杂,特别是两种聚合物中的双频吸收带非常明显。这些光谱有两个方面值得指出。首先,随着较重的硫族元素被取代到给体小分子中,整个双频带光谱被转移到较低的能量。第二,低能带比高能带偏移更大。此外,两种材料分别在300
‑
430nm,450
‑
780nm均有一吸收带,同时无论是在溶液中还是薄膜状态下这两种材料在波长450
‑
780nm之间都展现出强而宽的光响应,其中较短波长的峰可能归属于s和se原子取代的苯并二唑单元之间共轭产生的π
‑
π*跃迁吸收,而另一个峰可能是更高能量转换的结果。p1在氯苯溶液中的吸收峰值在695nm,p2吸收峰有10nm的红移,这与两种材料的分子给体单元的给电子能力相对应。相较于溶液状态,在薄膜状态下,p2膜显示出比p1更大的红移,暗示苯并硒二唑含有比苯并噻二唑更多的电子。同时,无论是p1膜还是p2膜,其吸收峰值比溶液吸收峰均有近20nm的红移,分别为705nm和730nm,且吸收更宽,这种红移可归结为固体状态下分子之间堆积所致。同时p2在635nm处有一较明显的肩峰,这说明p1在固体状态下呈现出更为有序的分子间聚集。p1和p2薄膜状态的边带吸收值分别为790nm和850nm。结果显示出可通过简单引入不同的杂原子来微调基于苯并二唑共轭单元共聚物的光物理性能,加上两种聚合物给体材料均呈现出宽的吸收(450
‑
780nm),说明这两种材料有望获得较为理想的光伏性能。
[0083]
实施例5、聚合物p1和p2的电化学性质
[0084]
通过循环伏安法(cv)测定p1和p2的电化学性质。采用盈思便携式电化学工作站,测试条件为:在室温,氩气保护下,电解液为浓度0.1mol/l的四丁基六氟磷酸铵(bu4npf6)的无水乙腈溶液,用标准的三电极电化学电池来测定,以铂碳电极为工作电极,铂丝为对电极,饱和甘汞电极为参比电极,扫描速度为60mv/s。从图中可以看出p1和p2的起始还原电位(e
oxon
)分别为
‑
0.35ev,
‑
0.3ev,起始氧化电位(e
oxon
)分别为1.1ev,1.17ev。可以由起始氧化电位(e
oxon
)和起始还原电位(e
redon
)通过相应公式计算出两种聚合物的最高占有分子轨道能级(homo)和最低未占有分子轨道能级(lumo):e
homo/lumo
=
‑
e(e
ox/redon
4.71)(ev),得出p1和p2的homo能级分别为
‑
5.06ev,
‑
5.01ev,lumo能级分别为
‑
3.61ev,
‑
3.65ev。相应地,p1和p2的电化学带隙(e
gcv
)由公式e
gcv
=e
lumo
‑
e
homo
计算得到,故二者的e
gcv
分别为1.45ev,1.36ev。从能级出发,这三种共聚物均可成为有机太阳能电池给体材料,并且通过比较研究,可以看出与p2相比,p1的homo能级更低,从而为p1获得较高的开路电压(v
oc
)提供保证。
[0085]
以上所述仅为本发明的较佳实施例,凡依本发明申请专利范围所做的均等变化与修饰,皆应属本发明的涵盖范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。