1.本发明属于有机合成领域,涉及一种苯乙酮的制备方法。
背景技术:
2.苯乙酮是一种重要的有机合成中间体,可用作合成香料、药物以及其他有机合成的原料,同时也是纤维素醚、纤维素酯和树脂的良好溶剂及塑料的增塑剂可用作制造香皂和纸烟,还可用作纤维素醚、纤维素酯、树脂、防腐剂、橡胶、医药、染料等的溶剂,市场需求很大,具有广阔的应用前景。
3.在传统的苯乙酮生产工艺中,主要是利用傅克烷基化均相反应,即在化学计量的无水三氯化铝催化下由苯与乙酰氯、乙酸酐或乙酸反应制取。这种方法产生大量的有毒且腐蚀性强的废液,易污染环境,催化剂用量大,重复利用及产物分离都较困难,基于这些缺点该方法受到了限制。
4.乙苯氧化法也是工业上经常用于生产苯乙酮的方法,该方法是以金属配合物为催化剂,在均相高温下氧化乙苯制备苯乙酮,当该催化体系转化率不高,副产物多。
5.此外,苯乙酮也可利用光化学反应制备,具体过程包括:将乙苯或α
‑
甲基苯乙烯在光催化剂作用下使用h2o2或有机过氧化物为氧化剂,在光照射下可得到苯乙酮,其中,光催化剂主要有钛硅分子筛、含铬分子筛,金属配合物等。但此种方法对催化剂和反应装置要求高,反应成本较为昂贵。
6.因此,开发一种收率高、生产成本低、对环境危害小的苯乙酮制备方法具有重要意义。
技术实现要素:
7.针对现有技术中存在的缺陷,本发明提供一种苯乙酮的制备方法,该制备方法以廉价易得的异丙苯、氧气、硫酸亚铁等为原料,通过简单的氧化、还原等反应过程,能以高选择性制备得到苯乙酮,且反应后生成的副产物碱式硫酸铁与甲醇也可回收利用,因此该制备方法具有收率高、经济效应好、反应成本低、环境友好、操作简单的优点。
8.本发明提供一种苯乙酮的制备方法,包括以下步骤:
9.1)在加热条件下,利用氧气对含有异丙苯的碱性溶液进行氧化处理,得到氧化反应液;
10.2)将硫酸亚铁溶液加入所述氧化反应液中进行反应,得到苯乙酮。
11.如上所述的制备方法,其中,所述碱性溶液中的碱的质量占所述异丙苯的质量的0.01~0.08%。
12.如上所述的制备方法,其中,所述碱性溶液选自氢氧化钠水溶液和/或碳酸钠水溶液。
13.如上所述的制备方法,其中,步骤1)中,加热温度为110~150℃。
14.如上所述的制备方法,其中,步骤1)中,所述氧气通入所述碱性溶液的流量为0.1
~0.6l/min。
15.如上所述的制备方法,其中,所述氧化处理依次包括第一氧化处理和第二氧化处理,所述第一氧化处理的时间为1h,所述第二氧化处理的时间为5h;
16.所述第一氧化处理中,氧气流量为0.25l/min;
17.所述第二氧化处理中,氧气流量为0.4l/min。
18.如上所述的制备方法,其中,步骤2)中,将所述硫酸亚铁溶液滴加入所述氧化反应液中,滴加速度为200~600g/min。
19.如上所述的制备方法,其中,所述硫酸亚铁与所述异丙苯的摩尔比为(1.0~2.0):1。
20.如上所述的制备方法,其中,步骤2)中,反应温度为10~15℃。
21.如上所述的制备方法,其中,步骤2)中,待硫酸亚铁溶液完全加入所述氧化反应液后,再保温反应10min。
22.本发明所提供的苯乙酮的制备方法,以廉价易得的异丙苯为起始原料,制备过程中所用到的试剂氧气、硫酸亚铁等都来源广泛、无毒无害,该方法通过简单的氧化、还原等反应过程,能够高选择性的制备得到苯乙酮,且反应后生成的副产物甲醇可作为溶剂回收利用,所生成的副产物碱式硫酸铁可作为絮凝剂应用于净水领域。因此该制备方法具有收率高经济效应好、反应成本低、环境友好、操作简单的优点,有望于应用于苯乙酮的工业化生产中。
附图说明
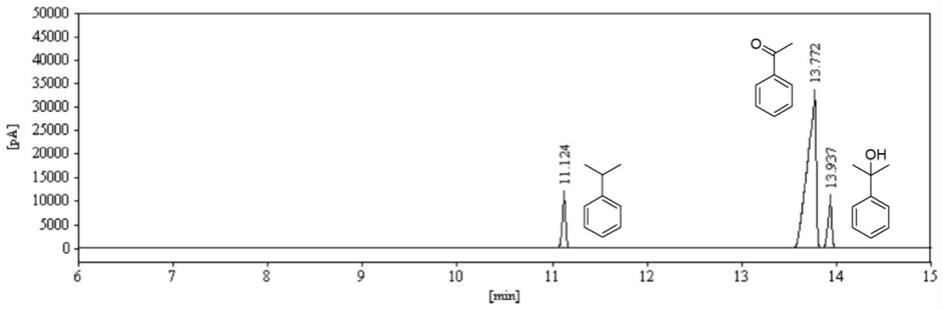
23.图1为实施例1的有机相gc分析图。
具体实施方式
24.为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明的实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
25.本发明提供一种苯乙酮的制备方法,包括以下步骤:
26.1)在加热条件下,利用氧气对含有异丙苯的碱性溶液进行氧化处理,得到氧化反应液;
27.2)将硫酸亚铁溶液加入氧化反应液中进行反应,得到苯乙酮。
28.上述制备方法可用如下反应式表示:
[0029][0030]
发明人推测该反应经历了如下的反应历程:
[0031]
步骤1)中,加热条件可引发异丙苯生成自由基中间体,自由基中间体与氧气反应后可进一步反应得到过氧化氢异丙苯;
[0032]
步骤2)中,硫酸亚铁作为还原剂,可对过氧化氢异丙苯进行还原,还原经历两个阶
段,首先,在硫酸亚铁作用下,过氧化氢异丙苯脱除一分子氢氧自由基,接着,再脱除一分子甲基自由基即可得到苯乙酮。
[0033]
在此过程中,氢氧自由基可与甲基自由基结合得到甲醇,生成的甲醇可分离后作为溶剂回收利用。而还原剂硫酸亚铁在经历还原反应后可转化为碱式硫酸铁(fe(oh)so4),碱式硫酸铁作为絮凝剂可用于净水领域中。
[0034]
此外,在还原的第一阶段,过氧化氢异丙苯在脱除一分子氢氧自由基得到2
‑
苯异丙氧基自由基中间体,该中间体夺取异丙苯上的一分子氢生成副产物2
‑
苯异丙醇。该制备方法通过使用氧气对含有异丙苯的碱性溶液进行氧化处理后,使用硫酸亚铁溶液再对氧化反应液进行还原,能够以不低于75%的选择性制得苯乙酮,同时可使副产物2
‑
苯异丙醇的选择性不高于25%。
[0035]
本发明苯乙酮的制备方法,以廉价易得的异丙苯作为起始原料,反应过程中所使用的辅助试剂氧气、硫酸亚铁等均为大宗原料,来源广泛,通过氧化、还原等反应过程,能够高选择性的制备得到苯乙酮,且反应后生成的甲醇、碱式硫酸铁都可回收利用。因此该制备方法具有收率高、成本低、经济效应好,环境友好的优点。
[0036]
碱性溶液在氧化反应的过程中主要起催化剂的作用,碱性溶液中的碱能够与异丙苯叔碳上的氢原子之间相互作用,降低异丙苯中叔碳的c
‑
h键离解能,提高自由基链增长的速度,促进异丙苯向过氧化氢异丙苯的转化。作为步骤1)反应的催化剂,碱性溶液中所加入的碱的质量占异丙苯的质量的0.01~0.08%时,能够达到较好的促进反应的效果。
[0037]
进一步的,碱性溶液可选自氢氧化钠水溶液和/或碳酸钠水溶液,示例性的,碱性溶液可以是氢氧化钠溶液,也可以是碳酸钠水溶液,或者氢氧化钠水溶液和碳酸钠水溶液的混合溶液。氢氧化钠和碳酸钠都是非常廉价易得的原料,能够进一步降低反应的成本。
[0038]
发明人在实验过程中发现,当碱性溶液进一步优选自碳酸钠水溶液时,反应具有更高的苯乙酮选择性。
[0039]
进一步的,步骤1)中,加热温度为110~150℃。加热是引发异丙苯热裂解得到自由基中间体的重要条件,上述加热温度范围能够在相对温和的条件下促进自由基氧化反应的进行。
[0040]
步骤1)中所使用的氧气并不局限于纯氧,只要是含氧气的气体即可,例如空气。
[0041]
当使用纯氧进行氧化时,在氧化前需先使用惰性气体对反应体系进行置换。惰性气体可选自氮气、氦气和氩气中的至少一种。
[0042]
可以理解的是,当异丙苯和氧气分别处于气液两相状态时,在加压条件下将更有利于氧化反应的进行。具体的,可以通过向反应体系中通入惰性气体使反应压力达到1.0~1.1mpa,在该压力范围内,氧化反应可顺利进行。
[0043]
在一种具体的实施方式中,可以通过控制氧气通入碱性溶液的流量为0.1~0.6l/min,使氧化反应的速度更加平稳可控,氧气流量太小,氧化反应难以进行,氧气流量太大,反应将会生成更多的副产物。
[0044]
进一步的,发明人研究发现,在反应过程中适当调节氧气通入碱性溶液的流量,使氧化处理分阶段进行将更有利于氧化反应的进行。例如,可使氧化处理依次包括第一氧化处理和第二氧化处理,第一氧化处理的时间为1h,第二氧化处理的时间为5h;其中,在第一氧化处理中,氧气流量为0.25l/min;在第二氧化处理中,氧气流量为0.4l/min。
[0045]
待步骤1)的反应完成后,得到的氧化反应液中包括过氧化氢异丙苯中间体,在步骤2)中,以硫酸亚铁为还原剂对氧化反应液进行还原,为控制氧化还原反应的速度,避免过度还原现象的发生,可将硫酸亚铁溶液滴加入氧化反应液中,并控制滴加速度为200~600g/min。
[0046]
进一步的,当控制硫酸亚铁与异丙苯的摩尔比为(1.0~2.0):1时,在有助于异丙苯转化率提升的同时,还能够避免还原剂的浪费。
[0047]
当硫酸亚铁溶液加入氧化反应液时,反应速度很快,会有大量热量放出,为避免反应速度过快产生不必要的副产物,可使反应在低温条件下进行,具体的,可控制反应温度为10~15℃。
[0048]
待硫酸亚铁溶液完全加入氧化反应液后,使反应体系再保温反应10min即可有助于反应进行的更加充分,有利于高选择性的得到苯乙酮产物。
[0049]
以下,将结合具体的实施例对本发明所提供的苯乙酮的制备方法进行进一步地介绍。在下述实施例中,如无特殊说明,所有原料均可通过商购或常规方法制备得到。
[0050]
实施例1
[0051]
本实施例苯乙酮的制备方法包括以下步骤:
[0052]
1)将异丙苯(1200g,10mol)加入到2l的高压釜中,再加入0.3g碳酸钠和100ml水,密闭,搅拌;
[0053]
2)向高压釜中通入氮气至1.0~1.1mpa,随后加热至110℃,通入氧气,并将尾气阀门打开,控制氧气的流量为0.25l/min,保持该流量一小时后,调节氧气流量至0.4l/min,继续反应5h后,停止反应,得到氧化反应液;
[0054]
3)将feso4·
7h2o(3336g,12mol)溶于33.36kg的水中,得到硫酸亚铁溶液,控制温度为10~15℃将硫酸亚铁溶液以400g/min的滴加速度加入氧化反应液中,滴加完毕后,继续保温反应10min;
[0055]
4)保温完成后,将反应液分液,有机相使用gc分析;
[0056]
gc的分析测试条件如下:
[0057]
采用安捷伦a7890气相色谱仪,检测器为fid,气化室温度为280.0℃,氢气流量为30.0ml/min,空气流量为400.0ml/min,采用氮气尾气吹扫,流量为25.0ml/min。
[0058]
图1为实施例1的有机相gc分析图,对图1进行处理后可得到表1中的相关信息:
[0059]
表1
[0060]
保留时间/min峰高/pa峰面积/(pa*s)峰面积%含量/%组分名称11.12410488.3227603.4710.339710.3397异丙苯13.77231839.41211116.3179.079779.0797苯乙酮13.9379641.3628246.8110.580710.58072
‑
苯异丙醇
[0061]
根据表1中的数据可以分析得出,本实施例的异丙苯的转化率为90%,苯乙酮的选择性为88%,副产物2
‑
苯异丙醇的选择性为12%。
[0062]
实施例2
[0063]
本实施例苯乙酮的制备方法包括以下步骤:
[0064]
1)将异丙苯(1200g,10mol)加入到2l的高压釜中,再加入0.5g碳酸钠和100ml水,密闭,搅拌;
[0065]
2)向高压釜中通入氮气至1.0~1.1mpa,随后加热至110℃,通入氧气,并将尾气阀门打开,控制氧气的流量为0.25l/min,保持该流量一小时后,调节氧气流量至0.4l/min,继续反应5h后,停止反应,得到氧化反应液;
[0066]
3)将feso4·
7h2o(3336g,12mol)溶于33.36kg的水中,得到硫酸亚铁溶液,控制温度为10~15℃将硫酸亚铁溶液以400g/min的滴加速度加入氧化反应液中,滴加完毕后,继续保温反应10min;
[0067]
4)保温完成后,将反应液分液,有机相使用gc分析,分析得出,异丙苯的转化率为92%,苯乙酮的选择性为80%,副产物2
‑
苯异丙醇的选择性为20%。
[0068]
实施例3
[0069]
本实施例苯乙酮的制备方法包括以下步骤:
[0070]
1)将异丙苯(1200g,10mol)加入到2l的高压釜中,再加入0.1g碳酸钠和100ml水,密闭,搅拌;
[0071]
2)向高压釜中通入氮气至1.0~1.1mpa,随后加热至110℃,通入氧气,并将尾气阀门打开,控制氧气的流量为0.25l/min,保持该流量一小时后,调节氧气流量至0.4l/min,继续反应5h后,停止反应,得到氧化反应液;
[0072]
3)将feso4·
7h2o(3336g,12mol)溶于33.36kg的水中,得到硫酸亚铁溶液,控制温度为10~15℃将硫酸亚铁溶液以400g/min的滴加速度加入氧化反应液中,滴加完毕后,继续保温反应10min;
[0073]
4)保温完成后,将反应液分液,有机相使用gc分析,分析得出,异丙苯的转化率为85%,苯乙酮的选择性为86%,副产物2
‑
苯异丙醇的选择性为14%。
[0074]
实施例4
[0075]
本实施例苯乙酮的制备方法包括以下步骤:
[0076]
1)将异丙苯(1200g,10mol)加入到2l的高压釜中,再加入0.3g氢氧化钠和100ml水,密闭,搅拌;
[0077]
2)向高压釜中通入氮气至1.0~1.1mpa,随后加热至110℃,通入氧气,并将尾气阀门打开,控制氧气的流量为0.25l/min,保持该流量一小时后,调节氧气流量至0.4l/min,继续反应5h后,停止反应,得到氧化反应液;
[0078]
3)将feso4·
7h2o(3336g,12mol)溶于33.36kg的水中,得到硫酸亚铁溶液,控制温度为10~15℃将硫酸亚铁溶液以400g/min的滴加速度加入氧化反应液中,滴加完毕后,继续保温反应10min;
[0079]
4)保温完成后,将反应液分液,有机相使用gc分析,分析得出,异丙苯的转化率为89%,苯乙酮的选择性为86%,副产物2
‑
苯异丙醇的选择性为14%。
[0080]
实施例5
[0081]
本实施例苯乙酮的制备方法包括以下步骤:
[0082]
1)将异丙苯(1200g,10mol)加入到2l的高压釜中,再加入0.5g氢氧化钠和100ml水,密闭,搅拌;
[0083]
2)向高压釜中通入氮气至1.0~1.1mpa,随后加热至110℃,通入氧气,并将尾气阀门打开,控制氧气的流量为0.25l/min,保持该流量一小时后,调节氧气流量至0.4l/min,继续反应5h后,停止反应,得到氧化反应液;
[0084]
3)将feso4·
7h2o(3336g,12mol)溶于33.36kg的水中,得到硫酸亚铁溶液,控制温度为10~15℃将硫酸亚铁溶液以400g/min的滴加速度加入氧化反应液中,滴加完毕后,继续保温反应10min;
[0085]
4)保温完成后,将反应液分液,有机相使用gc分析,分析得出,异丙苯的转化率为70%,苯乙酮的选择性为75%,副产物2
‑
苯异丙醇的选择性为25%。
[0086]
实施例6
[0087]
本实施例苯乙酮的制备方法包括以下步骤:
[0088]
1)将异丙苯(1200g,10mol)加入到2l的高压釜中,再加入0.3g碳酸钠和100ml水,密闭,搅拌;
[0089]
2)向高压釜中通入氮气至1.0~1.1mpa,随后加热至110℃,通入氧气,并将尾气阀门打开,控制氧气的流量为0.25l/min,保持该流量一小时后,调节氧气流量至0.4l/min,继续反应5h后,停止反应,得到氧化反应液;
[0090]
3)将feso4·
7h2o(4170g,15mol)溶于44.70kg的水中,得到硫酸亚铁溶液,控制温度为10~15℃将硫酸亚铁溶液以400g/min的滴加速度加入氧化反应液中,滴加完毕后,继续保温反应10min;
[0091]
4)保温完成后,将反应液分液,有机相使用gc分析,分析得出,异丙苯的转化率为93%,苯乙酮的选择性为88%,副产物2
‑
苯异丙醇的选择性为12%。
[0092]
实施例7
[0093]
本实施例苯乙酮的制备方法包括以下步骤:
[0094]
1)将异丙苯(1200g,10mol)加入到2l的高压釜中,再加入0.3g碳酸钠和100ml水,密闭,搅拌;
[0095]
2)向高压釜中通入氮气至1.0~1.1mpa,随后加热至150℃,通入氧气,并将尾气阀门打开,控制氧气的流量为0.25l/min,保持该流量一小时后,调节氧气流量至0.4l/min,继续反应5h后,停止反应,得到氧化反应液;
[0096]
3)将feso4·
7h2o(4170g,15mol)溶于44.70kg的水中,得到硫酸亚铁溶液,控制温度为10~15℃将硫酸亚铁溶液以400g/min的滴加速度加入氧化反应液中,滴加完毕后,继续保温反应10min;
[0097]
4)保温完成后,将反应液分液,有机相使用gc分析,分析得出,异丙苯的转化率为93%,苯乙酮的选择性为88%,副产物2
‑
苯异丙醇的选择性为12%。
[0098]
实施例8
[0099]
本实施例苯乙酮的制备方法包括以下步骤:
[0100]
1)将异丙苯(1200g,10mol)加入到2l的高压釜中,再加入0.3g碳酸钠和100ml水,密闭,搅拌;
[0101]
2)向高压釜中通入氮气至1.0~1.1mpa,随后加热至150℃,通入氧气,并将尾气阀门打开,控制氧气的流量为0.25l/min,保持该流量一小时后,调节氧气流量至0.4l/min,继续反应5h后,停止反应,得到氧化反应液;
[0102]
3)将feso4·
7h2o(4170g,15mol)溶于44.70kg的水中,得到硫酸亚铁溶液,将硫酸亚铁溶液加入氧化反应液中,在10~15℃下混合反应1h。
[0103]
4)反应完成后,分液,分离的有机相使用gc分析,通过分析得出,异丙苯的转化率
为93%,苯乙酮的选择性为85%,副产物2
‑
苯异丙醇的选择性为15%。
[0104]
实施例9
[0105]
本实施例苯乙酮的制备方法包括以下步骤:
[0106]
1)将异丙苯(1200g,10mol)加入到2l的高压釜中,再加入0.3g碳酸钠和100ml水,密闭,搅拌;
[0107]
2)向高压釜中通入氮气至1.0~1.1mpa,随后加热至150℃,通入氧气,并将尾气阀门打开,控制氧气的流量为0.4l/min,保持该流量反应6h后,停止反应,得到氧化反应液;
[0108]
3)将feso4·
7h2o(4170g,15mol)溶于44.70kg的水中,得到硫酸亚铁溶液,控制温度为10~15℃将硫酸亚铁溶液以400g/min的滴加速度加入氧化反应液中,滴加完毕后,继续保温反应10min;
[0109]
4)保温完成后,将反应液分液,有机相使用gc分析,分析得出,异丙苯的转化率为88%,苯乙酮的选择性为80%,副产物2
‑
苯异丙醇的选择性为20%。
[0110]
以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。