提高目的片段与psicheck2载体连接效率的方法
技术领域
1.本发明涉及一种提高目的片段与psicheck2载体连接效率的方法,属于分子生物学技术领域。
背景技术:
2.据了解,基因工程研究包括四大步骤,即目的基因的获取,基因表达载体的构建,将基因表达载体转入受体细胞,目的基因的监测与鉴定,这其中最重要的也是最繁琐的莫过于基因表达载体的构建。载体构建是分子生物学与基因工程中的一种重要的实验技术,是研究基因功能的必要手段,该技术一般要求较高的重组效率和准确性。目前常用的载体有质粒载体、噬菌体载体和柯斯质粒,其中质粒载体在动物实验中应用最为广泛。质粒载体源于细菌,并且外源性质粒可以导入dh5α感受态细菌,通过培养细菌实现质粒的大规模提取,目前已有多种商业化质粒提取试剂盒。质粒可以在其他物种细胞中自我复制,并且其dna序列中包含丰富的核酸内切酶位点。这些特性使得质粒载体在分子生物学和基因工程中得到广泛应用。
3.psicheck2载体是一种质粒载体,它可监测与报告基因融合的目的基因表达的变化。该载体应用海肾萤光素酶作为主要报告基因,目的片段被克隆至海肾萤光素酶翻译终止密码子下游的多克隆位点。由合成的sirna 或体内表达的shrna 引发的针对目的基因的rnai过程,导致融合mrna 的剪切和随后的降解。通过检测海肾萤光素酶活性的变化即可判定sirna 或体内表达的shrna与目的片段是否存在靶向关系。该载体目前在研究microrna与基因3’utr互作实验中具有重要意义。psicheck2载体构建现在有两种方法,一种是常规引物法,另一种是同源重组法。目前通常采用常规引物法,即保护碱基 酶切位点 目的片段序列,但是该方法由于保护碱基较短,会导致其pcr产物双酶切效率大大降低,因此实验的连接效率很低。而同源重组法的构建过程相对简单,可以省去很多的时间和精力,但是假阳性率略高。虽然目前已有商业化同源重组载体构建试剂盒可以降低假阳性率,但是费用昂贵。
技术实现要素:
4.为了克服现有技术的不足,本发明的目的包括但不限于提供一种提高目的片段与psicheck2载体连接效率的方法。
5.为了实现上述目的,本发明采用了以下技术方案:本发明提供一种提高目的片段与psicheck2载体连接效率的方法,该方法的步骤如下:以牛基因组dna为模板,设计载体同源序列替换传统引物中的保护碱基,利用pcr扩增目的片段,将pcr扩增产物与psicheck2载体连接后导入感受态细胞,于37℃空气摇床培养2小时后,涂板,再于37℃恒温培养箱培养16小时,对单克隆计数,并随机挑取单克隆,进行菌液pcr和sanger测序,判断目的片段与psicheck2载体的载体重组效率。
6.本发明的方法是以牛基因组dna为模板,利用载体同源序列替换常规引物中的保
护碱基,通过pcr扩增牛igfbp3基因部分片段,将pcr产物与psicheck2经核酸内切酶xho i和not i处理后,利用t4连接酶连接,连接产物导入dh5α感受态细胞,最后根据细菌单克隆数目、琼脂糖凝胶电泳结果和测序结果,判断目的片段与psicheck2载体重组效率。
7.本发明进一步优化的技术方案如下:优选地,所述目的片段为牛igfbp3基因部分片段,位于牛igfbp3基因候选区域chr4: 76123453
‑
76124382,其核苷酸序列如seq id no.1所示,psicheck2载体的核苷酸序列如seq id no.2所示。
8.优选地,所述载体重组效率是按照细菌单克隆数目、菌液pcr阳性率和测序正确率,判断目的片段与psicheck2载体的连接效率。
9.优选地,所述所述用于扩增牛igfbp3基因部分片段的引入载体同源片段引物对p1为:上游引物f1(其核苷酸序列如seq id no.3所示):5
′‑
taggcgatcgctcgaggcacaaaagactgccaaggaca
ꢀ‑3′
,下游引物r1(其核苷酸序列如seq id no.4所示):5
′‑ꢀ
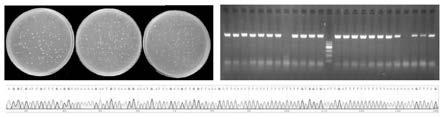
ttgcggccagcggccgccaccaagcaagggcgatt
ꢀ‑3′
;所述常规扩增引物对p2为:上游引物f2(其核苷酸序列如seq id no.5所示):5
′‑ꢀ
ccgctcgaggcacaaaagactgccaagga
ꢀ‑3′
,下游引物r2(其核苷酸序列如seq id no.6所示):5
′‑ꢀ
ataagaatgcggccgccaccaagcaagggcgatttt
ꢀ‑3′
。
10.优选地,所述的载体同源序列引物中的载体同源序列为:上游引物载体同源序列:taggcgatcg下游引物载体同源序列:ttgcggcca;所述的常规引物中保护碱基为:上游引物保护碱基:ccg下游引物保护碱基:ataagaat。
11.优选地,所述基于引物对p1的pcr扩增产物的片段大小为963 bp,基于引物对p2的pcr扩增产物的片段大小为955 bp。
12.优选地,所述基于引物对p1的pcr扩增产物、psicheck2载体经核酸内切酶xho i和not i处理,再经t4连接酶连接后,导入dh5α感受态细胞,记为同源片段实验组。
13.基于引物对p1的pcr扩增产物和经核酸内切酶xho i和not i处理后的psicheck2载体直接导入dh5α感受态细胞,记为细菌内源性同源重组试验组。
14.基于引物对p2的pcr扩增产物、psicheck2载体经核酸内切酶xho i和not i处理,再经t4连接酶连接后,导入dh5α感受态细胞,记为常规引物试验组。
15.优选地,所述pcr所用的扩增体系包括50 ng/μl模板dna 1.0 μl,10 mm引物对p1或p2所对应的上下游引物各1.0 μl,2
×
mastermix(sangon)12.5 μl,以及去离子水9.5 μl;所述pcr的反应程序为:95 ℃预变性5min;94 ℃变性30 s,60 ℃退火30 s,72 ℃延申60 s共35循环。
16.本发明还提供上述方法在重组载体构建中的应用。
17.上述的应用,是利用同源序列引物重组载体的菌单克隆数目、菌液pcr阳性率和测序正确率要高于常规引物重组载体和细菌内源性同源重组载体,即同源片段实验组的连接效率要比常规引物实验组和细菌内源性同源重实验组效率高,可为其他载体构建提供技术依据。
18.本发明中,以牛igfbp3基因chr4: 76123453
‑ꢀ
76124382作为扩增区域。利用两组引物分别进行pcr扩增,其中同源序列引物为载体同源片段 酶切位点 目的片段序列,常规引物为保护碱基 酶切位点 目的片段序列。扩增产物进行不同处理后导入dh5α,其中同源片段实验组为双酶切p1 pcr 双酶切psicheck2载体 t4连接,细菌内源性同源重组实验组为p1 pcr 双酶切psicheck2载体,常规引物实验组为双酶切p2 pcr产物和双酶切psicheck2载体 t4连接。载体重组效率按照菌单克隆数目、菌液pcr阳性率和测序正确率结果,判断目的片段与psicheck2载体的重组效率。
19.本发明并不是基于同源重组原理,而是通过改变保护碱基的长度(本发明中采用的是载体同源序列)提高双酶切效率达到改善重组效率的目的。目前商业化的核酸内切酶,其酶切效率受保护碱基数目的影响。基于传统引物扩增的pcr产物其xho
ꢀⅰꢀ
的2小时酶切效率仅有10%,not
ꢀⅰ
的2小时酶切效率为25%。并且双酶切存在“木桶效应”,即使某个酶切效率达到100%,如果另一个酶切效率很低,同样会造成载体连接效率低下。采用同源序列引物法构建psicheck2载体,该方法对比常规引物法、同源重组法(非试剂盒)的连接效率,发现同源序列引物法的菌单克隆数目、菌液pcr阳性率和测序正确率均高于其他两种方法。
20.与现有技术相比,本发明具有以下优点:(1)本发明中同源片段实验组连接效率要比常规引物实验组和细菌内源性同源重实验组效率高;(2)该方法方法准确可靠、操作简单、成本低;(3)为其他载体构建提供技术依据。
附图说明
21.图1为本发明中同源片段实验组单克隆图片、菌液pcr和测序结果图。
22.图2为本发明中细菌内源性同源重组实验组单克隆图片、菌液pcr和测序结果图。
23.图3为本发明中常规引物实验组单克隆图片和菌液pcr结果图。
具体实施方式
24.下面结合实施例对本发明的技术方案做进一步的详细说明:本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护权限不限于下述的实施。下列实施例中未注明具体条件的实验方法,按照本领域常规方法和条件,或商品说明书选择。下列实施例所涉及的试剂及材料均为市购,此处不一一列举。
25.本发明所涉及的pcr是以牛基因组dna为模板,以牛igfbp3基因候选区域chr4: 76123453
‑ꢀ
76124382为扩增区域,利用两组引物分别进行pcr扩增,其中同源序列引物设计为载体同源片段 酶切位点 目的片段序列,常规引物为保护碱基 酶切位点 目的片段序列。扩增产物进行不同处理后导入dh5α,其中同源片段实验组为双酶切p1 pcr 双酶切psicheck2载体 t4连接,细菌内源性同源重组实验组为p1 pcr 双酶切psicheck2载体,常
规引物实验组为双酶切p2 pcr产物和双酶切psicheck2载体 t4连接。载体重组效率按照菌单克隆数目、菌液pcr阳性率和测序正确率结果,判断目的片段与psicheck2载体的重组效率。
26.实施例11、牛样本采集以牛作为检测对象,从陕西秦宝牧业有限公司采集2只牛颈静脉血液样本。
27.2、基因组dna的分离、提取、纯化参考sambrock et al (2002)方法。
28.3、目标序列及内参序列的扩增以ncbi数据库(http://www.ncbi.nlm.nih.gov/)公布的牛igfbp1基因序列为参考序列,利用primer 5.0设计扩增igfbp3扩增引物,依据xho i和not i酶切位点在引物5’端添加酶切序列和保护碱基或者酶切序列和载体同源片段。引物对序列信息如表1所示。
29.表1 pcr引物信息pcr所用的扩增体系以25 μl计为:pcr所用的扩增体系包括50 ng/μl模板dna 1.0 μl,10 mm引物对p1或p2所对应的上下游引物各1.0 μl,2
×
mastermix(sangon)12.5 μl,以及去离子水9.5 μl。。
30.进行pcr所用的的反应程序为:95 ℃预变性5min;94 ℃变性30 s,60 ℃退火30 s,72 ℃延申60 s共35循环。
31.4、pcr产物的处理同源片段实验组是基于p1引物的pcr扩增产物和psicheck2载体均经酸内切酶xho i和not i处理后,经t4连接酶连接后,导入dh5α感受态细胞。
32.细菌内源性同源重组实验组是基于p1引物的pcr扩增产物和经核酸内切酶xho i和not i处理后的psicheck2载体直接导入dh5α感受态细胞。
33.常规引物实验组是基于p2引物pcr扩增产物和psicheck2载体均利用核酸内切酶xho i和not i处理后,经t4连接酶连接后,导入dh5α感受态细胞。
34.5、载体连接效率判定对同源片段实验组、细菌内源性同源重组实验组、常规引物实验组转化后的dh5α分别进行涂板,每组3个板子,统计其单克隆个数,结果见图1至图3。利用spss 18软件单因素方差分析判定每组单克隆个数差异。
35.每个板子随机挑取8个单克隆,不足8个的全部挑取,利用各自对应的p1或p2引物进行菌液pcr扩增,统计三个实验组菌液pcr阳性率,结果见图1至图3。利用spss 18软件卡方检验判定每组菌液pcr阳性率差异。
36.每个板子重新随机挑取8个单克隆,不足8个的全部挑取,进行测序,确定序列正确率,结果见图1至图3。利用spss 18软件卡方检验判定每组连接序列正确性差异。
37.分析结果如表2所示,同源片段实验组和细菌内源性同源重组实验组的单克隆数、菌液pcr阳性率、序列正确性均显著高于常规引物实验组,并且同源片段实验组的序列正确性显著高于细菌内源性同源重组实验组,表明该发明可以显著提高重组载体的构建成功率。
38.表2igfbp3基因片段与psicheck2载体连接效率分析6、上述载体构建方法在分子生物学中的应用本发明中同源片段实验组连接效率要比常规引物实验组和细菌内源性同源重实验组效率高,该方法方法准确可靠、操作简单、成本低,同时可为其他载体构建提供技术依据。
39.以上所述,仅为本发明中的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉该技术的人在本发明所揭露的技术范围内,可理解想到的变换或替换,都应涵盖在本发明的包含范围之内,因此,本发明的保护范围应该以权利要求书的保护范围为准。。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。