用于多重拷贝数变异检测和等位基因比率定量的定量扩增子测序
1.相关申请的交叉引用
2.本技术要求2019年1月4日提交的美国临时申请号62/788,375的优先权权益,该申请的全部内容通过引用并入本技术。
3.关于联邦政府赞助的研究的声明
4.本发明是在美国国立卫生研究院授予的政府拨款号r01hg008752的支持下完成的。政府对这项发明享有一定的权利。
5.序列表的参考
6.本技术包含序列表,该序列表已通过efs
‑
web以ascii格式提交,并且在此以其全部内容通过引用并入。所述ascii副本创建于2019年11月26日,命名为ricep0058wo_st25.txt,大小为145.6kb。
技术领域
7.本发明总体上涉及分子生物学和医学领域。更具体地,它涉及使用定量扩增子测序,进行多重拷贝数变异检测和等位基因比率定量的组合物和方法。
背景技术:
8.拷贝数变异(cnv)是重要的癌症生物标志物,导致癌症的形成和进展。它们存在于很大比例的肿瘤中,根据癌症类型介于3%到98%之间。许多cnv赋予靶定疗法敏感性或抗性,例如,met扩增使得对非小细胞肺癌中met tki的敏感性增加,而pten缺失赋予黑色素瘤中的braf抑制剂抗性。在肿瘤样品中,由于肿瘤异质性和正常细胞污染,特定基因的cnv可能仅存在于一小部分(<10%)细胞中。
9.与突变和插入缺失不同,cnv没有唯一序列,因此检测cnv需要准确定量。由于dna分子采样的随机性,这种定量很困难。例如,每个基因座采样1200个分子(即来自600个正常细胞的1200个单倍体基因组拷贝,4ng基因组dna)的标准偏差(σ)可以通过泊松分布估计:细胞的1200个单倍体基因组拷贝,4ng基因组dna)的标准偏差(σ)可以通过泊松分布估计:对应分子数量的3%。在这种情况下,不可能检测到1%的额外拷贝。理论上,增加输入分子的数量或分析更多的基因座可以同样降低方差,并且σ可以估计为如果基因组拷贝数或基因座数增加100倍,σ将减少到0.3%,并且可以检测到1%的额外拷贝。
10.目前分子诊断中cnv检测的标准方法是原位杂交(ish),它可以基于对少量细胞的观察来确定cnv状态。然而,由于荧光和明视野显微镜中可区分的颜色数量有限,ish技术缺乏同时分析多个基因组区域的能力。此外,ish是一个复杂的过程,需要由专门的实验室执行,因此无法被广泛采用。
11.cnv检测的另一种方法是液滴数字pcr(ddpcr),这是一种基于pcr的dna分子绝对定量方法。然而,通过大量重复实验,它对cnv的检测限(lod)是大约20%的额外拷贝。与ish
一样,由于荧光通道数量有限,ddpcr也无法进行多重检测。基于微阵列的方法,包括阵列比较基因组杂交和snp阵列,是用于筛选大型cnv和非整倍体的高度多重检测方法。然而,它们在检测<40kb的较小cnv或额外拷贝<30%的低频cnv方面表现不佳。
12.下一代测序(ngs)是一种高通量技术,在过去十年中成本迅速降低。ngs在癌症分子诊断领域很受欢迎。已在ngs平台上实现并商业化了lod<0.1%变体等位基因频率的高度多重突变检测。然而,当前用于cnv检测的ngs方法的lod并不那么好:全外显子组测序(wes)已用于额外拷贝的水平约为30%的cnv发现,但价格昂贵,并且需要更多的ngs读段(成本成比例增加)以实现更低的lod。较小的杂交捕获组合(panel),例如foundationone商业组合,可以以较低的成本达到约30%的额外拷贝的lod。
13.在用于诊断的ngs组合中,需要富集靶标以减少在无关基因组区域上浪费的ngs读段。两种流行的靶标富集方法是杂交捕获和多重pcr。当前基于ngs的cnv组合大多基于杂交捕获,这意味着靶标区域由生物素化核酸探针捕获,并使用链霉亲和素磁珠与基因组的其余部分分离。当组合尺寸较小时,杂交捕获组合的在靶率较低,因此大多数组合>100kb(即>1000个探针或基因座);这是由于不想要的dna在珠子表面、探针和捕获的靶标上发生非特异性结合。由于基因座数量众多,杂交捕获组合的覆盖率并不统一:95%和5%的百分位基因座相差至少30倍,这在定量中引入了另一层偏差。由于末端修复和连接不完善,杂交捕获组合还存在转化率低(即测序输入分子的百分比)的问题,从而导致采样处理有偏差并导致变异。
技术实现要素:
14.本文提供了定量扩增子测序的方法,用于通过聚合酶链式反应用寡核苷酸条形码序列标记dna样品中靶定基因组基因座的每条链,并扩增用于高通量测序的基因组区域。通过定量每个基因的额外拷贝的频率,这些方法可用于同时检测一组目标基因中的拷贝数变异(cnv)。此外,这些方法使用多重pcr对靶定基因组基因座的不同遗传同一性的等位基因比率进行定量。
15.在一个实施例中,本文提供了用于制备用于高通量测序的基因组dna靶定区域的方法,该方法包括:(a)获取基因组dna样品;(b)通过使用如下进行两个pcr循环来扩增至少一部分基因组dna样品:(i)第一寡核苷酸,其从5
′
到3
′
包括第一区域、长度为0至50个核苷酸的第二区域(例如,0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49或50个核苷酸)、包含至少四个简并核苷酸的第三区域(例如,4、5、6、7、8、9、10、11或12个简并核苷酸)、以及包含与第一靶标基因组dna区域互补的序列的第四区域;以及(ii)第二寡核苷酸,其从5
′
至3’包括第五区域、长度为0至50个核苷酸的第六区域(例如,0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49或50个核苷酸)和以及包含与第二靶标基因组dna区域互补的序列的第七区域;(c)使用比步骤(b)中所使用的退火温度高0
‑
10℃(例如,1
‑
10、2
‑
10、3
‑
10、4
‑
10、5
‑
10、1
‑
9、1
‑
8、1
‑
7、1
‑
6、1
‑
5、2
‑
9、2
‑
8、2
‑
7℃或其中可导出的任何范围或值)的退火温度,并使用如下进行至少三个pcr循环来扩增步骤(b)的产物:(i)第三寡核苷酸,其包括能够与第一区域的至少一部分的反向互补序列杂交
的序列;以及(ii)第四寡核苷酸,其包括能够与第五区域的至少一部分的反向互补序列杂交的序列;以及(d)通过使用第五寡核苷酸进行至少一个pcr循环来扩增步骤(c)的产物,该第五寡核苷酸从5
′
到3
′
包括第八区域、长度在0和50个核苷酸之间的第九区域(例如,0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49或50个核苷酸),以及包含与第三靶标基因组dna区域互补的序列的第十区域,其中第三靶标基因组dna区域比第二靶标基因组dna区域更靠近第一靶标基因组dna区域至少一个核苷酸。
16.在一些方面,方法是用于制备用于高通量测序的基因组dna的1至10,000个靶定区域(例如,至少1、2、3、4、5、6、7、8、9、10、15、20、25、30、35、40、45、50、75、100、250、500、750、1000、2000、3000、4000或5000和至多10,000、9000、8000、7000、6000、5000、4000、3000、2000、1000、750、500、250、100、75或50个靶定区域,或其中可导出的任何范围或值)的方法。在一些方面,第三区域是唯一分子标识符(umi)。在一些方面,第三靶标基因组dna区域比第二靶标基因组dna区域更靠近第一靶标基因组dna区域1
‑
10个(例如,1、2、3、4、5、6、7、8、9或10个)碱基。在一些方面,第一区域和第八区域是通用引物结合位点。在一些方面,第一区域和第八区域包含完整或部分ngs衔接子序列。在一些方面,第五区域包含在人类基因组中找不到的序列。在一些方面,第五区域包含不同于ngs衔接子序列的序列。在一些方面,第一区域和第五区域的解链温度为0
‑
10℃(例如,1
‑
10、2
‑
10、3
‑
10、4
‑
10、5
‑
10、1
‑
9、1
‑
8、1
‑
7、1
‑
6、1
‑
5、2
‑
9、2
‑
8、2
‑
7℃或其中可导出的任何范围或值)高于第四区域和第七区域的解链温度。在一些方面,第三区域中的简并核苷酸各自独立地是a、t或c中的一个。在一些方面,第三区域中的简并核苷酸都不是g。在一些方面,存在第一寡核苷酸群体,每个都有唯一的第三区域。
17.在一些方面,该方法进一步包括纯化步骤(c)的产物。在一些方面,纯化包括spri纯化或柱纯化。在一些方面,该方法进一步包括纯化步骤(d)的产物。在一些方面,纯化包括spri纯化或柱纯化。在一些方面,该方法进一步包括(e)使用与第一区域和第八区域杂交的引物通过pcr扩增步骤(d)的产物,其中该引物包含用于下一代测序的索引序列。在一些方面,该方法进一步包括纯化步骤(e)的产物。在一些方面,纯化包括spri纯化或柱纯化。在一些方面,该方法进一步包括(f)对步骤(e)的产物进行高通量dna测序。在一些方面,高通量dna测序包括下一代测序。
18.在一些方面,第一靶标基因组dna区域和第二靶标基因组dna区域在基因组dna的相反的链上。在一些方面,第一靶标基因组dna区域和第二靶标基因组dna区域相隔40个核苷酸至500个核苷酸(例如,40、45、50、55、60、65、70、75、80、90、100、125、150、175、200、225、250、275、300、325、350、375、400、425、450、475或500个核苷酸,或其中可导出的任何值)。在一些方面,步骤(b)包括约30分钟(例如27、28、29、30、31、32或33分钟)的延伸时间。在一些方面,步骤(c)包括约30秒(例如27、28、29、30、31、32或33秒)的延伸时间。在一些方面,步骤(d)包括约30分钟(例如27、28、29、30、31、32或33分钟)的延伸时间。
19.在一个实施例中,本文提供了用于定量至少一个靶标基因的额外拷贝的频率(fec)的方法,该方法包括:(a)获取基因组dna样品;(b)根据本发明实施例中任一项的方法制备用于高通量测序的基因组dna,其中第四区域、第七区域和第十区域的序列与至少一个靶标基因杂交;(c)根据本实施例中任一项的方法进行高通量测序;以及(d)基于步骤(c)中
获得的测序信息计算至少一个靶标基因的fec。
20.在一些方面,该方法是定量一组靶标基因的fec的方法,其中该组靶标基因包含2至1000个靶标基因(例如,至少2、3、4、5、6、7、8、9、10、15、20、25、30、35、40、45、50、75、100、250、500或750,最多1,000、900、800、750、700、650、600、550、500、450、400、350、300、250、200、150、100、75、50、25、20、15、10、9、8、7、6、5、4或3个靶定区域,或任何其中可导出的范围或值)。在一些方面,使用第一寡核苷酸群体、第二寡核苷酸群体和第五寡核苷酸群体进行步骤(b),其中第一、第二和第五寡核苷酸群体中的每一个群体的一部分分别包含与该组靶标基因中的一个互补的第四、第七和第十区域。在一些方面,第四、第七和第十区域中的每一个都包含在人类基因组中仅发现一次的序列。在一些方面,与一个靶标基因杂交的每个第一寡核苷酸和与相同靶标基因杂交的每个其它第一寡核苷酸相比,具有唯一第三区域。在一些方面,使用分别包含与参考基因互补的第四、第七和第十区域的第一寡核苷酸、第二寡核苷酸和第五寡核苷酸进行步骤(b)。在一些方面,步骤(b)制备用于高通量测序的每个靶标基因或参考基因的一部分,其中该部分长度在40个核苷酸和500个核苷酸之间(例如,以40、45、50、55、60、65、70、75、80、90、100、125、150、175、200、225、250、275、300、325、350、375、400、425、450、475或500个核苷酸,或其中可导出的任何值)。在一些方面,fec定义为:
[0021][0022]
在一些方面,步骤(d)包括:(i)将ngs读段与每个靶标基因的靶定部分进行比对,并基于它们比对的基因座将ngs读段分组到亚组中;(ii)在每个基因座基于其umi序列划分ngs读段,以便将所有携带相同umi序列的ngs读段分组为一个umi家族;(iii)去除因pcr错误或ngs错误得到的umi家族;(iv)计算每个基因座上唯一umi序列的数量;以及(v)基于每个靶标基因和参考基因中每个基因座的唯一umi序列的数量计算fec。在一些方面,步骤(d)(iii)包括去除不符合umi简并碱基设计的umi序列。在一些方面,步骤(d)(iii)包括去除umi家族规模小于fmin的umi家族,其中umi家族规模是携带相同umi的读段的数量,其中fmin在2和20之间(例如,2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20)。在一些方面,步骤(d)(iv)包括去除与具有更大家族规模的另一个umi序列仅相差一个或两个碱基的umi序列。
[0023]
在一些方面,fec定义为:
[0024][0025]
其中是全部或部分所述靶标基因基因座的唯一umi数量的总和,u是要考虑的基因座数,u不超过所述靶标基因中的基因座总数;是全部或部分参考基因座的唯一umi数量的总和,v是一个参考的要考虑的基因座数,v不超过所述参考中的基因座的总数;w是要考虑的参考数,w不超过参考总数;且k由实验校准确定。在一些方面,fec用于鉴定靶标基因的拷贝数变异(cnv)状态。
[0026]
在一个实施例中,本文提供了用于定量至少一个靶标基因组基因座的不同遗传同一性的等位基因比率的方法,该方法包括:(a)获取基因组dna样品;(b)根据本发明实施例中任一项的方法制备用于高通量测序的基因组dna,其中第四区域、第七区域和第十区域的序列与至少一个靶标基因组基因座附近的基因组dna杂交;(c)根据本实施例中任一项的方
法进行高通量测序;以及(d)基于步骤(c)中获得的测序信息上计算至少一个靶标基因组基因座的不同遗传同一性的等位基因比率。
[0027]
在一些方面,该方法是用于定量一组靶标基因组基因座的不同遗传同一性的等位基因比率的方法,其中该组靶标基因组基因座包含2至10,000个靶标基因组基因座(例如,至少2、3、4、5、6、7、8、9、10、15、20、25、30、35、40、45、50、75、100、250、500、750、1,000、2,000、3,000、4,000或5,000和最多10,000、9,000、8,000、7,000、6,000、5,000、4,000、3,000、2,000、1,000、750、500、250、100、75或50个靶标基因组基因座,或任何其中可导出的范围或值)。在一些方面,使用第一寡核苷酸群体、第二寡核苷酸群体和第五寡核苷酸群体进行步骤(b),其中第一、第二和第五寡核苷酸群体中的每个群体的一部分分别包含与所述一组靶标基因组基因座中的至少一个附近的基因组dna互补的第四、第七和第十区域。在一些方面,第四、第七和第十区域中的每个包括在步骤(b)的条件下不能与基因组dna的非靶标区域杂交的序列。在一些方面,与一个靶标基因组基因座附近的基因组dna杂交的每个第一寡核苷酸和与相同靶标基因组基因座附近的基因组dna杂交的每个其它第一寡核苷酸相比,具有唯一第三区域。在一些方面,每个靶标基因组基因座的长度在40个核苷酸和500个核苷酸之间(例如,以40、45、50、55、60、65、70、75、80、90、100、125、150、175、200、225、250、275、300、325、350、375、400、425、450、475或500个核苷酸,或其中可推导出的任何值)。
[0028]
在一些方面,步骤(d)包括:(i)将ngs读段与靶定基因组基因座进行比对,并基于它们比对的基因座将ngs读段分组到亚组中;(ii)在每个基因座基于其umi序列划分ngs读段,以便将所有携带相同umi序列的ngs读段分组为一个umi家族;(iii)去除因pcr错误或ngs错误得到的umi家族;(iv)调用每个剩余umi家族的遗传同一性;(v)计算每个基因座上唯一umi序列的数量;以及(vi)计算等位基因比率。在一些方面,步骤(d)(iii)包括去除不符合umi简并碱基设计的umi序列。在一些方面,步骤(d)(iii)包括去除umi家族规模小于fmin的umi家族,其中umi家族规模是携带相同umi的读段的数量,其中fmin在2和20之间(例如,2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20)。在一些方面,步骤(d)(iii)包括去除与具有更大家族规模的另一个umi序列仅相差一个或两个碱基的umi序列。在一些方面,步骤(d)(iv)包括仅当的umi家族的至少70%(例如,70%、75%、80%、85%、90%、95%或98%)的读段在目标遗传基因座上相同时,调用遗传同一性。在一些方面,等位基因比率定义为r
allele
=n1/n2,其中n1是第一遗传同一性的唯一umi数量,n2是第二遗传同一性的唯一umi数量。
[0029]
在一些方面,步骤(d)(iv)包括鉴定每个umi家族的共有序列。在一些方面,共有序列是在umi家族中出现次数最多的序列。在一些方面,步骤(d)(iv)进一步包括将所述共有序列与所述基因座的野生型序列进行比较,从而鉴定所述共有序列中的突变。在一些方面,该方法进一步包括计算所鉴定突变的变体等位基因频率(vaf)。在一些方面,所鉴定突变的vaf定义为具有所述突变的umi家族的数量/umi家族的总数。
[0030]
如本文中所使用的,就特定组分而言,“基本上不含”在本文中被用于表示未将任何特定组分故意配制成组合物和/或仅作为污染物或微量存在。因此,由组合物的任何意外污染而产生的特定组分的总量远低于0.05%,优选低于0.01%。最优选的是使用标准分析方法无法检测特定组分的组合物。
[0031]
如本说明书所使用的,“a”或“an”可指一个或多个。如在一个或多个权利要求中所
使用的,当与“包含”一词一起使用时,“a”或“an”可指一个或多个。
[0032]
尽管本公开支持仅提及替代品和“和/或”的定义,在权利要求书中使用的术语“或”是指“和/或”,除非明确指出仅提及替代品或替代品相互排斥。如本文中所使用的“另一个”可表示至少第二个或更多。
[0033]
在整个应用中,术语“关于”用于指示包括装置的固有误差变化、用于确定值的方法或研究对象之间存在的变化的值。
[0034]
通过下面详细的描述,本发明的其他目的、特点和优点将会变得显而易见。然而应当理解,这些详细说明和特定的实施例在指示本发明的优选实施方案时,只是用于示范,因为根据此详细说明,在本发明的精神和范围内的各种变化和改进对于本领域技术人员将显而易见。
附图说明
[0035]
以下附图构成本说明书的一部分并被包括,以进一步说明本发明的某些方面。通过参考一个或多个附图,并结合此处给出的具体实施例的描述,可以更好地理解本发明。
[0036]
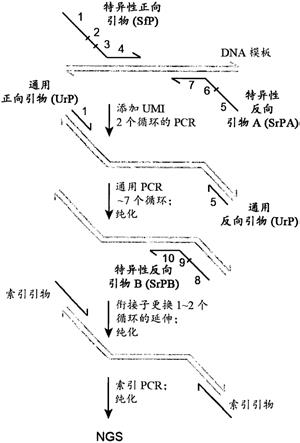
图1.qaseq引物设计和实验工作流程示意图.每个引物组含有3种不同的寡核苷酸:特异性正向引物(sfp)、特异性反向引物a(srpa)和特异性反向引物b(srpb)。每个qaseq组合只需要一个通用正向引物(ufp)和一个通用反向引物(urp)。在ufp或urp中,区域1或区域5的5
′
端可以有另外的碱基。对于推荐的工作流程,首先将dna样品与所有sfp、srpa、dna聚合酶、dntp和pcr缓冲液混合。执行2个循环的长延伸pcr以在所有靶标基因座上添加umi。接下来,为了在扩增分子的同时防止在同一原始分子上添加多个umi,在使用ufp和urp(短延伸,约30s)进行约7个循环的pcr扩增中将退火温度升高约8℃;请注意,将ufp和urp添加到反应中是热循环仪上的一个开管(open
‑
tube)步骤。使用spri磁珠或柱纯化后,将srpb引物、dna聚合酶、dntp和pcr缓冲液与pcr产物混合进行衔接子更换;经过2个循环的长延伸(约30min)后,ngs衔接子仅添加到正确的pcr产物上,而不是引物二聚体或非特异性产物上。在使用spri磁珠或柱再次纯化后,进行标准的ngs索引pcr;文库被归一化并加载到illumina测序仪上。
[0037]
图2.umi交叉结合能的模拟.使用(h)
20
代替(n)
20
or(sww)6sw作为umi序列会降低平均交叉结合能,表明引物
‑
二聚体相互作用较少。这里对每个umi模式进行了500次模拟;在每次模拟中,随机生成与模式一致的2个序列,并假设60℃和0.18m k
来计算这些序列之间的交叉结合δg
°
。
[0038]
图3a
‑
b.引物和umi之间的间隔区减少了pcr偏差.(图3a)评估引物和umi之间的间隔区的重要性的工作流程。将三组引物,即无间隔区(第1组);正向引物和umi之间有5nt间隔区,反向引物和umi之间有5nt间隔区(第2组);或正向引物和umi之间有12nt间隔区,反向引物和umi之间有11nt间隔区(第3组)分别用于扩增输入分子。在illumina miseq进行ngs分析之前添加了索引。(图3b)三组引物的实验性umi家族规模分布直方图。去除了与umi设计模式不匹配的umi序列。
[0039]
图4a
‑
b.基于umi的cnv绝对定量的数据分析.(图4a)cnv检测的数据分析工作流程。分析fastq输出文件中的ngs读段以生成cnv状态作为结果。靶标基因的fec将计算为
其中是全部或部分靶标基因基因座的唯一umi数量的总和,u是要考虑的基因座数;是全部或部分参考基因座的唯一umi数量的总和,v是一个参考要考虑的基因座数;w是要考虑的参考数;且k由实验校准确定。cnv状态基于fec确定。(图4b)数据分析中umi家族规模和唯一umi数量的定义:umi家族规模是携带相同umi序列的读段数量,唯一umi数量是一个基因座上不同umi序列的总数。
[0040]
图5.实验性umi家族规模分布的实例.同一ngs文库中10个erbb2扩增子和10个参考扩增子的umi家族规模分布实例。我们使用正常细胞系gdna na18562(购自coriell)作为20
‑
重qaseq实验的模板输入;输入样品包含2500个单倍体基因组拷贝。通过illumina miseq reagent kit v3(150次循环)使用150万个读段对制备的ngs文库进行测序。接受和丢弃的umi的分数显示为饼图。在所有umi中,约有20%因pcr或测序错误(即在poly(h)umi中发现g碱基)而被丢弃;约有40%因家庭规模小(≤3)被丢弃。
[0041]
图6.不同基因座的实验唯一umi数量的实例.每个基因座的唯一umi数量实例,对应于图5所示的数据;白条是erbb2扩增子,灰条是参考扩增子。输入样品包含2500个单倍体基因组拷贝。通过illumina miseq reagent kit v3(150次循环)使用150万个读段对制备的ngs文库进行测序。
[0042]
图7.正常细胞系gdna na18562的实验校准结果和模拟的理论标准偏差极限.cnv比率(σ
cnv比率
)的标准偏差针对输入分子数绘制。lod可以近似为3σ
cnv比率
。我们对每个不同的输入量(75、250、750和2500个单倍体基因组拷贝)进行了5次重复实验;实验结果绘制为十字符号。假设采样分子数的泊松分布进行了模拟;由于采样的随机性,模拟的σ
cnv比率
(绘制为虚线)是理论下限。
[0043]
图8a
‑
c.ffpe样品上cnv检测的实验结果的实例.我们测试了来自同一肿瘤的2个肺癌ffpe载玻片,其中不太可能发生erbb2 cnv。输入提取的dna样品包含每个ngs文库的2500个单倍体基因组拷贝。通过illumina miseq reagent kit v3(150次循环)使用150万个读段对制备的ngs文库进行测序。(图8a)针对扩增子erbb2_1和参考_1绘制了的umi家族规模分布实例;接受和丢弃的umi的分数显示为饼图。(图8b)每个扩增子区域的唯一umi数量实例。白条是erbb2扩增子;灰条是参考扩增子。(图8c)绘制了来自同一肺癌肿瘤的2张ffpe载玻片的cnv比率。基于先前的校准数据,使用qaseq在这些ffpe载玻片中未检测到erbb2的cnv。平均值和lod=3σ
cnv比率
是基于750个基因组拷贝输入细胞系gdna文库的数据计算的(参见图7),它们具有与ffpe样品相似的唯一umi数量。
[0044]
图9a
‑
e.使用主要实验工作流程减少引物二聚体.(图9a)我们测试过的最简单的工作流程是一锅反应:添加umi后,将索引引物直接添加到反应中作为热循环仪上的开管步骤,然后进行索引pcr(即通用pcr)。此工作流程的在靶率低(0.5%);脱靶ngs读段主要是引物二聚体。(图9b)6个循环的通用pcr后加入spri纯化步骤以减少引物二聚体;在靶率提高到20%。(图9c)索引pcr后增加了使用琼脂糖凝胶的尺寸选择步骤,以进一步减少引物二聚体;与图9b相比,在靶率有所提高,但仍低于50%。(图9d)主要实验工作流程包括衔接子更换和通用pcr后的纯化,平均在靶率高达66%。(图9e)工作流程9a
‑
d中引物二聚体的来源。
[0045]
图10a
‑
c.不需要ngs索引pcr的工作流程实例.(图10a)将索引和p5序列添加到ufp的5
′
;其他索引和p7序列添加到srpb的5
′
。从衔接子更换中获得的扩增子包含p5、p7和双索
引,因此已准备好进行测序。(图10b)将索引和p7序列添加到srpb的5
′
,并在衔接子更换步骤中将索引引物与srpb一起添加。扩增子已准备好进行测序。(图10c)将索引和p5序列添加到sfp的5
′
;带有p5序列的引物用作通用pcr步骤中的ufp。其他索引和p7序列添加到srpb的5
′
。扩增子已准备好进行测序。
[0046]
图11.qaseq引物设计和工作流程的变体.每个引物组含有3种不同的寡核苷酸:特异性正向引物(sfp)、特异性反向引物a(srpa)和特异性反向引物b(srpb)。与原始设计相比,srpa只需要模板结合区域,不需要通用反向引物(urp)。每个qaseq组合只需要一个通用正向引物(ufp);ufp中区域1的5
′
端可以有另外的碱基。与原来的实验工作流程相比,通用pcr步骤需要更多的pcr循环;建议≥10个循环。
[0047]
图12a
‑
b.基于qaseq的等位基因比率定量的数据分析.(图12a)等位基因比率定量的数据分析工作流程。分析fastq输出文件中的ngs读段以生成不同遗传同一性之间的等位基因比率。每个靶定基因座的等位基因比率计算为r
等位基因
=n1/n2,其中n1是第一遗传同一性的唯一umi数量,n2是第二遗传同一性的唯一umi数量。(图12b)基于多数票的每个umi家族的遗传同一性调用。
[0048]
图13.掺入临床ffpe样品的cnv检测实验结果实例.将两个先前表征的ffpe dna样品(1个“正常”样品和1个“erbb2扩增异常”样品)混合以生成2.5%、5%和10%的erbb2 fec样品。“正常”样品的erbb2 fec为0%,“erbb2扩增异常”样品的erbb2 fec为78%。将实验归一化fec值针对预期的erbb2 fec作图。“正常”样品重复测试5次,100
‑
重cnv组合的lod估计为“正常”样品的3个标准偏差。成功检测到2.5%、5%和10%erbb2 fec样品中的cnv,因为它们计算的fec超出了3个标准偏差范围。
[0049]
图14.使用qaseq进行突变定量的生物信息学工作流程.显示的是突变定量的数据处理工作流程的概要。
[0050]
图15.观察到的179
‑
重全面组合的分子数.输入为8.3ng(5000预期分子数)的100%multiplex i wild type cfdna reference standard(horizon discovery)。转化率平均为62%;该组合中的97%具有>10%的转化率。
[0051]
图16.179
‑
重全面组合的错误率.输入是8.3ng的100%multiplex i wild type cfdna reference standard(horizon discovery);同一样品一式三份进行测试。绘制了3840个不同基因座(使用umi纠错后)的错误率。3次重复的最高错误率为0.23%、0.20%和0.23%,平均错误率为0.006%、0.005%和0.005%。
[0052]
图17.179
‑
重全面组合的突变定量结果.使用的样品是0.3%cfdna参考标准(通过混合来自horizon discovery的0.1%multiplex i cfdna reference standard和1%multiplex i cfdna reference standard来创建),一式三份进行测试。6个突变的实验vaf与预期的vaf大体一致;差异主要是由于对少量(≤9)突变分子进行抽样的随机性。
具体实施方式
[0053]
本文提供了定量扩增子测序的方法,用于通过聚合酶链反应用寡核苷酸条形码序列标记原始dna样品中靶定基因组基因座的每条链,并扩增用于高通量测序的基因组区域。本文还提供了,允许通过对每个基因的额外拷贝的频率进行定量来同时检测一组目标基因中的拷贝数变异(cnv)的方法。所公开的方法还提供了,使用多重pcr对靶定基因组基因座
的不同遗传同一性的等位基因比率的定量。这些方法可用于检测肿瘤样品中目标基因的cnv,指导靶定疗法的选择,并帮助了解癌症的形成和进展。
[0054]
目前单基因疾病产前诊断的标准方法是对从侵入性和危险性绒毛膜绒毛取样或羊膜穿刺术中获得的胎儿遗传物质进行测序。单基因疾病的遗传无创产前检测(nipt)基于母体血浆中胎儿来源的无细胞dna(cfdna)的循环。由于背景母体dna的存在,可信地检测由胎儿cfdna引起的等位基因比率变化变得具有挑战性,尤其是当母体dna在目标基因座上是杂合的时。液滴数字pcr(ddpcr)已被用于定量携带致病突变的突变体等位基因与nipt的野生型等位基因之间的等位基因比率(lun et al.,2008),但实际可行性受到技术精度和可靠性的限制。qaseq通过向原始输入分子的每条链添加唯一分子标识符来实现dna分子的绝对定量,并可应用于nipt的等位基因比率定量。因此,qaseq也可用于等位基因比率定量。等位基因比率定量旨在定量具有不同遗传同一性的dna分子的比率。准确的等位基因比率定量是单基因疾病(如β
‑
地中海贫血和囊性纤维化)的nipt的关键。
[0055]
i.cnv额外拷贝的频率
[0056]
基因组dna样品中cnv的额外拷贝的频率(fec)在本文中定义为:
[0057][0058]
fec的正值表示样品中靶标基因组区域的扩增,fec的负值表示样品中靶标基因组区域的缺失。
[0059]
虽然qaseq可用于定量fec,但它不提供有关肿瘤组织样品中含有cnv的细胞的百分比的信息。例如,如果肿瘤样品中1%的细胞含有4个erbb2拷贝,而其余99%的细胞含有2个拷贝,则fec为1%;如果样品中0.5%的细胞含有6个erbb2拷贝,其余99.5%的细胞含有2个拷贝,则fec仍为1%。此外,qaseq不提供有关额外拷贝的基因组位置的信息。
[0060]
ii.多重pcr组合设计
[0061]
在qaseq多重pcr组合中,一个靶标基因需要m(m=1~1000)组引物,每组引物扩增靶标基因区域中的一个非重叠小区域(40nt至500nt,通常≤200nt)。如果该组合有多个靶标基因,则用于每个基因的引物组数量相似(≈m)。该组合还含有相似数量(≈m)的引物组,用于扩增参考基因组区域。参考基因座用作加载的基因组dna(gdna)量的内标,因此不需要对样品中的dna浓度进行准确定量。每个组合可以使用至少一个参考引物组。因为增加靶标基因中输入分子或基因座的数量都可以减少随机采样的变化,所以每个基因可以使用更多数量的引物组来改善含有较少dna的样品类型的lod;在这种情况下,需要按比例增加参考引物组的数量。
[0062]
每个引物组含有三种不同的寡核苷酸:特异性正向引物(sfp)、特异性反向引物a(srpa)和特异性反向引物b(srpb)(参见图1)。sfp从5
′
到3
′
包括区域1、2、3和4。区域4是模板结合区域;区域3是umi;区域1是完整或部分ngs衔接子;区域2是可选添加的间隔区(通常为0~15nt),用于均匀扩增umi。srpa从5
′
到3
′
包括区域5、6和7。区域7是模板结合区域;区域5是用于通用扩增的定制衔接子(即与ngs衔接子不同且在人类基因组中找不到的序列);区域6是可选添加的间隔区(通常为0~15nt),用于均匀扩增不同基因座。srpb从5
′
到3
′
包括区域8、9和10。区域10是模板结合区,其3’端比区域7更靠近区域4至少1个碱基;区域8是完整或部分ngs衔接子;区域9是可选添加的间隔区(通常为0~15nt),用于均匀扩增不同基
因座。每个qaseq组合只需要一个通用正向引物(ufp)和一个通用反向引物(urp)。ufp包含区域1,urp包含区域5;ufp或urp中区域1或区域5的5
′
端可以有另外的碱基。模板结合区域4、7、10的解链温度(tm)与pcr退火温度大致相同,实验pcr条件下ufp和urp的tm不低于4、7、10区。
[0063]
在设计引物时,应避免在引物结合区域出现具有显著次要等位基因频率(maf)的单核苷酸多态性(snp),这样引物的结合亲和力就不会受到不同患者样品中核苷酸序列变体的影响。此外,应搜索全人类基因组核苷酸序列,以确保引物不易发生非靶标区域的非特异性扩增。
[0064]
在靶向肿瘤样品的福尔马林固定石蜡包埋(ffpe)标本中erbb2的cnv的组合实例中,在erbb2基因区域设计了10组引物,每组引物扩增60至70nt的扩增子。此外,还设计了10组参考引物,每组都扩增来自不同染色体的不同管家基因中的区域(表1)。引物是使用matlab代码自动设计的,以满足上述设计原则,同时最大限度地减少引物相互作用。此外,避免了群体中maf>0.2%的非致病性snp。在线工具primer
‑
blast用于确保每个引物组在人类基因组中只有一个扩增子。引物序列见表2。
[0065]
表1.扩增子的位置
[0066]
扩增子名称染色体基因erbb21~10chr.17erbb2参考1chr.1psmb2参考2chr.3rpl32参考3chr.5rack1参考4chr.6tbp参考5chr.9vcp参考6chr.11hmbs参考7chr.12naca参考8chr.15b2m参考9chr.19gpi参考10chr.20top1
[0067]
表2.示例性qaseq组合中的引物序列
[0068]
[0069]
[0070]
[0071][0072]
表3.179
‑
重全面组合中的引物序列
[0073]
[0074]
[0075]
[0076]
[0077]
[0078]
[0079]
[0080]
[0081]
[0082]
[0083]
[0084]
[0085]
[0086]
[0087]
[0088]
[0089]
[0090]
[0091]
[0092]
[0093]
[0094]
[0095]
[0096]
[0097]
[0098]
[0099]
[0100]
[0101]
[0102]
[0103]
[0104][0105]
iii.umi设计
[0106]
在ngs文库制备过程中,pcr扩增步骤会显著增加定量变化,从而难以区分原始分子数的微小变化。umi技术可用于减少pcr偏差并实现原始dna分子的绝对定量。umi的概念是给每一个原始dna分子一个不同的dna序列作为“条形码”,这样每个ngs读段的来源都可以基于条形码序列进行追踪。给定足够的ngs读段,在ngs输出中发现的唯一umi的数量可以反映原始dna分子的数量。此前,umi技术主要用于基于ngs的低频突变检测中的纠错;它也被应用于定量。通过使用大量不同的umi序列来唯一地标记每个原始分子;例如,对100,000个原始分子使用109个不同的umi序列将产生<0.006%的携带重复umi的分子。
[0107]
含有简并碱基的dna序列,例如poly(n)(即在每个位置的a、t、c或g的混合物),通常用作umi序列。在qaseq中,poly(h)(a、t或c)用作umi,因为与poly(n)相比或与s(c或g)和w(a或t)碱基的混合相比,其具有较弱的交叉结合能,如模拟所示(图2)。(h)
20
包含3.5
×
109个不同的序列,其对于作为输入的100,000个分子是足够的;(h)
15
包含1.4
×
107个不同的序列,其对于作为输入的6,000个分子是足够的。
[0108]
iv.用以减少pcr偏差的间隔区
[0109]
pcr效率因不同序列的扩增子而异。由于umi由许多不同的序列组成,引物和可变umi区域之间的间隔区可用于实现更均匀的pcr效率。
[0110]
进行ngs以评估间隔区对pcr偏差的影响(图3a)。模板分子在5’端和3’端有两个用于扩增的衔接子,在中间有由(d)
15
组成的umi区域。将三组引物,即无任何间隔区(第1组);正向引物和umi之间有5nt间隔区,反向引物和umi之间有5nt间隔区(第2组);或正向引物和umi之间有12nt间隔区,反向引物和umi之间有11nt间隔区(第3组)分别用于扩增模板。在ngs分析之前通过pcr添加索引。(d)
15
含有1.4
×
107个不同的序列。由于输入模板分子数远低于可能的序列数量,每个唯一umi序列在扩增前只有1个拷贝。携带相同umi的所有ngs读段假定来自相同的分子。因此,umi家族规模(即携带相同umi的读段数量)是pcr效率的指标。
[0111]
比较umi家族规模分布以评估间隔区对pcr偏差的显著性(图3b)。当引物和umi之间的间隔区较长时,观察到更均匀的分布。在引物组3中,其中两端的间隔区长度都长于10nt,实现了显著改善的分布。
[0112]
v.qaseq工作流程
[0113]
qaseq ngs文库制备工作流程示意图示于图1.首先,将dna样品与所有sfp、srpa、
dna聚合酶、dntp和pcr缓冲液混合。执行两个循环的长延伸(约30min)pcr以在所有靶标基因座上添加umi。之后,一个dna分子中的每条链都将携带不同的umi。接下来,扩增分子时为了防止将多个umi添加到同一原始分子上,将退火温度升高约8℃,并使用ufp和urp用短延长(约30秒)进行至少两个循环(例如,大约七个循环)的扩增。将ufp和urp添加到反应中是热循环仪上的一个开管步骤。使用spri磁珠或柱纯化后,将srpb引物、dna聚合酶、dntp和pcr缓冲液与pcr产物混合进行衔接子更换;经过至少一个循环(例如两个循环)的长延伸(约30min)后,ngs衔接子仅添加到正确的pcr产物上,而不是引物二聚体或非特异性产物上。在使用spri磁珠或柱再次纯化后,进行标准的ngs索引pcr;文库被归一化并加载到illumina测序仪上。
[0114]
可以使用所有类型的dna聚合酶和pcr超混合物。应遵循所用具体聚合酶的标准退火、延伸和变性温度(通用pcr步骤除外,其中退火温度升高)。
[0115]
vi.替代性qaseq工作流程
[0116]
可以使用sfp和srpb执行工作流程,用两个循环的pcr添加umi,然后直接添加用于索引pcr的索引引物。为了测试这一点,在相同反应中使用了二十组sfp和srpb。该方法的实验在靶率非常低(0.5%),因此该方法可能不适用于诊断的ngs测定(图9a)。脱靶ngs读段主要是引物二聚体。在第二个替代性工作流程中,使用ufp和urp进行通用pcr进行六个循环的通用pcr,然后是纯化步骤。这些另外的步骤将不同文库的在靶率提高到12
‑
28%(平均在靶率=20%)(图9b)。测试了基于第二个替代性工作流程的第三个替代性工作流程。为此,在索引pcr后添加了使用琼脂糖凝胶的尺寸选择步骤,以进一步减少引物二聚体。实验平均在靶率提高到42%,但仍低于50%(图9c)。使用主要实验工作流程实现引物二聚体减少,其中包括通用pcr后的衔接子更换和纯化,并导致66%的高平均在靶率(图9d)。上述工作流程中引物二聚体的一个来源显示在图9e。如果sfp的3
′
部分与sfpb结合,或sfpb的3
′
部分与sfp结合,则可以生成在5
′
和3
′
端具有通用区域的二聚体链,从而在通用或索引pcr步骤中进行扩增。
[0117]
主要工作流程包括最终的索引pcr步骤,以将索引序列和测序仪的p5/p7序列添加到扩增子的末端;然而,有在umi添加、通用pcr或衔接子更换步骤期间添加上述序列的替代性工作流程,因此不需要索引pcr步骤。图10a
‑
c示出了三个实例。首先,将索引和p5序列添加到ufp的5
′
;将其他索引和p7序列添加到srpb的5
′
。从衔接子更换中获得的扩增子含有p5、p7和双索引,因此已准备好进行测序(图10a)。其次,将索引和p7序列添加到srpb的5
′
,并且在衔接子更换步骤中将此修饰的srpb与正常的p5索引引物混合(图10b)。第三,将索引和p5序列添加到sfp的5’;带有p5序列的引物用作通用pcr步骤中的ufp。将其他索引和p7序列添加到srpb的5
′
(图10c)。
[0118]
一种替代性qaseq引物设计和工作流程示于图11.每个引物组含有三种不同的寡核苷酸:特异性正向引物(sfp)、特异性反向引物a(srpa)和特异性反向引物b(srpb)。sfp从5
′
到3
′
包括区域1、2、3和4。区域4是模板结合区域;区域3是umi;区域1是完整或部分ngs衔接子;区域2是可选添加的间隔区(0~15nt),用于均匀扩增umi。srpa包含区域5,它是模板结合区域。srpb从5
′
到3
′
包括区域6、7和8。区域8是模板结合区,其3’端比区域5更靠近区域4至少1个碱基;区域6是完整或部分ngs衔接子;区域7是可选添加的间隔区(0~15nt),用于均匀扩增不同基因座。每个qaseq组合只需要一个通用正向引物(ufp),其包含区域1;ufp中
区域1的5
′
端可以有另外的碱基。模板结合区域4、5、8的解链温度(tm)与pcr退火温度大致相同,实验pcr条件下ufp的tm不低于区域4、5、8。与原始设计相比,srpa只需要模板结合区域,不需要通用反向引物(urp)。在实验工作流程中,在这种替代性引物设计下,通用pcr步骤中需要更多的pcr循环(例如,至少10个循环)。
[0119]
vii.数据分析工作流程
[0120]
cnv检测的数据分析工作流程示意图示于图4a中示出。首先,将原始ngs读段与扩增子区域比对;在比对之前可以进行可选的衔接子修整。丢弃未对准的读段,将对准了的读段按它们比对的基因座分组。
[0121]
然后,用umi序列进一步划分所有与相同基因座比对的读段,即将携带相同umi的读段分组为一个umi家族。umi家族规模是携带相同umi的读段数量,唯一umi数量是一个基因座上不同umi序列的总数(图4b)。接下来,去除所有可能是pcr或ngs错误结果的唯一umi家族。例如,与设计的umi模式不一致的umi序列(例如,在poly(h)umi序列中发现的g碱基)是错误的,应该去除。此外,如果两个umi序列仅相差1
‑
2个碱基,则具有较小umi家族规模的一个家族可能突变自另一个家族,因此可以任选地去除。去除umi错误后,也去除家族规模<f
min
的umi家族。f
min
是基于umi家族规模的分布确定的,大多数情况下可以使用f
min
=4。去除umi后的唯一umi数量(n)用于下一步骤。
[0122]
靶标基因的fec可以计算为:
[0123][0124]
其中是全部或部分所述靶标基因基因座的唯一umi数量的总和,u是要考虑的基因座数,u不超过所述靶标基因中的基因座总数;是全部或部分参考基因座的唯一umi数量的总和,v是一个参考的要考虑的基因座数,v不超过所述参考中的基因座的总数;w是要考虑的参考数,w不超过参考总数;且k由实验校准确定。在临床样品上测试qaseq组合之前,对具有充分表征的靶标基因cnv状态的dna样品进行了校准实验。从具有通过ddpcr表征的cnv状态的正常和癌细胞系中提取的gdna可用于校准。正常校准样品的fec应为0。测定的lod也由校准实验确定;lod是测定可检测到的额外拷贝的最小频率。在测试临床样品时,目标基因的fec将用于推断cnv状态;如果fec>lod,则推断样品含有靶标基因的扩增;如果fec≤lod,则推断样品含有靶标基因的缺失。
[0125]
viii.等位基因比率定量
[0126]
qaseq可用于使用多重pcr对1~10,000个基因组基因座的不同遗传同一性的等位基因比率进行定量。靶定基因组基因座的多重pcr组合设计,以及通过pcr用寡核苷酸条形码序列标记靶定基因组基因座的每条链,然后扩增基因组区域以进行高通量测序的实验工作流程与cnv检测类似。
[0127]
等位基因比率定量的数据分析工作流程示意图显示于图12a中示出。首先,将原始ngs读段与扩增子区域比对;在比对之前可以进行可选的衔接子修整。丢弃未对准的读段,将对准了的读段按它们比对的基因座分组。在每个基因座上,ngs读段除以umi序列;所有携带相同umi序列的ngs读段都分组为一个umi家族。如数据分析工作流程章节所述,去除了umi中存在错误的唯一umi家族,这些错误可能是pcr或ngs错误的结果。
[0128]
基于多数票调用每个剩余的umi家族的遗传同一性(野生型或突变型);遗传同一
性需要同一umi家族中至少70%的成员(读段)支持。以图12b为例,对于umi家族规模=7的umi家族,所有7个读段共享相同的umi序列(显示为2d条码)。目标基因座上的遗传同一性,6个读段是“a”,1个读段是“g”。由于umi家族中超过70%的读段支持“a”,因此该umi家族的遗传同一性称为“a”。对应于“g”的1个读段是pcr或ngs错误的结果。没有超过70%读段支持一种共用遗传同一性的umi家族被丢弃。
[0129]
接下来,针对靶定基因座上的每个不同遗传同一性计算唯一umi数量n(一个基因座上不同umi序列的总数);n表示原始链数量。靶标基因座的等位基因比率计算为r
等位基因
=n1/n2,其中n1是第一遗传同一性的唯一umi数量,n2是第二遗传同一性的唯一umi数量。
[0130]
ix.定义
[0131]
如本文所用,“扩增”是指用于增加一个或多个核苷酸序列的拷贝数的任何体外方法。核酸扩增导致核苷酸掺入dna或rna。如本文所用,一个扩增反应可以由多轮dna复制组成。例如,一个pcr反应可以包含30个至100个变性和复制“循环”。
[0132]“聚合酶链式反应”或“pcr”是指通过dna的互补链的同时引物延伸在体外扩增特定dna序列的反应。换句话说,pcr是制备侧接引物结合位点的靶核酸多拷贝或复制的反应,这种反应包括以下步骤的一个或多个重复:(i)使靶核酸变性,(ii)将引物退火至引物结合位点,和(iii)在三磷酸核苷存在下通过核酸聚合酶延伸引物。通常,反应在热循环仪器中通过每个步骤优化的不同温度循环。特定的温度、每个步骤的持续时间,以及步骤之间的变化率取决于本领域普通技术人员众所周知的许多因素,例如以参考文献为例:mcpherson等人,编辑,pcr:a practical approach and pcr2:a practical approach(irl press,oxford,1991 and 1995,respectively)。
[0133]“引物”指天然或合成的寡核苷酸,当与某一多核苷酸模板形成一个双链体后,即能作为核酸合成的一个起始点并且从其3
′
末端沿着模板延伸,从而形成一个延伸的双链体。延伸过程中所添加的核苷酸序列取决于模板多核苷酸的序列。通常,引物通过dna聚合酶延伸。引物的长度通常与其在引物延伸产物的合成中的使用相容,并且长度通常在介于8个至100个核苷酸之间的范围,例如长度在介于10个至75个、15个至60个、15个至40个、18个至30个、20个至40个、21个至50个、22个至45个、25个至40个等,更通常在18个至40个、20个至35个、21个至30个核苷酸之间的范围,以及在所述范围之间的任何长度。通常的引物的长度可以在介于10个至50个核苷酸之间的范围,例如15个至45个、18个至40个、20个至30个、21个至25个等,以及介于所述范围之间的任何长度。在一些实施例中,引物的长度通常不大于约10个、12个、15个、20个、21个、22个、23个、24个、25个、26个、27个、28个、29个、30个、35个、40个、45个、50个、55个、60个、65个或70个核苷酸。
[0134]
如本文所用,“掺入”是指成为核酸聚合物的一部分。
[0135]
如本文所用,术语“在不存在外源操纵的情况下”是指在不改变核酸分子被修饰的溶液的情况下对核酸分子进行修饰。在具体实施例中,它在没有手动操作或没有改变溶液条件的机器的情况下发生,这也可以称为缓冲条件。但是,在修饰过程中可能会发生温度变化。
[0136]“核苷”是碱基糖的组合,即缺少磷酸的核苷酸。在本领域中所公认的是,在使用术语核苷和核苷酸方面存在一定的互换性。例如,核苷酸脱氧尿苷三磷酸,即dutp,是脱氧核糖核苷三磷酸。掺入dna后,它作为dna单体,形式上是脱氧尿苷酸,即dump或单磷酸脱氧尿
苷。可以说,即使所得的dna中没有dutp部分,也可以将dutp掺入dna中。类似地,可以说,即使只是底物分子的一部分,也可以将脱氧尿苷掺入dna中。
[0137]
如本文所用,“核苷酸”是本领域的术语,是指碱糖磷酸的组合。核苷酸是核酸聚合物,即dna和rna的单体单元。该术语包括核糖核苷酸三磷酸,例如ratp、rctp、rgtp或rutp,和脱氧核糖核苷酸三磷酸,例如datp、dctp、dutp、dgtp或dttp。
[0138]
术语“核酸”或“多核苷酸”通常是指dna、rna、dna
‑
rna嵌合体或其衍生物或类似物的至少一个分子或链,所述至少一个分子或链包含至少一个核碱基,诸如dna中发现的天然存在的嘌呤或嘧啶碱基(例如,腺嘌呤“a”、鸟嘌呤“g”、胸腺嘧啶“t”和胞嘧啶“c”)或rna(例如,a、g、尿嘧啶“u”和c)中发现的天然存在的嘌呤或嘧啶碱基。术语“核酸”涵盖术语“寡核苷酸”和“多核苷酸”。如本文所用,“寡核苷酸”统称为且可互换地称为两个本领域术语,“寡核苷酸”和“多核苷酸”。请注意,尽管寡核苷酸和多核苷酸是不同的技术术语,但它们之间没有确切的分界线,它们在本文中可互换使用。术语“衔接子”也可以与术语“寡核苷酸”和“多核苷酸”互换使用。此外,术语“衔接子”可以表示线性衔接子(单链或双链)或茎环衔接子。这些定义通常是指至少一个单链分子,但是在特定的实施例中,还将涵盖与该至少一个单链分子部分地、基本上或完全互补的至少一条附加链。因此,核酸可涵盖至少一个双链分子或至少一个三链分子,该至少一个双链分子或至少一个三链分子包含构成该分子的链的特定序列的一条或多条互补链或“互补序列”。如本文所用,单链核酸可以用前缀“ss”表示,双链核酸用前缀“ds”表示,三链核酸用前缀“ts”表示。
[0139]“核酸分子”或“核酸靶标分子”是指任何单链或双链核酸分子,包括标准经典碱基、超修饰碱基、非天然碱基或其碱基的任何组合。例如但不限于,核酸分子含有四种经典dna碱基
‑
腺嘌呤、胞嘧啶、鸟嘌呤和胸腺嘧啶,和/或四种经典rna碱基
‑
腺嘌呤、胞嘧啶、鸟嘌呤和尿嘧啶。当核苷含有2
′‑
脱氧核糖基团时,尿嘧啶可以替代胸腺嘧啶。核酸分子可以从rna转化为dna,也可以从dna转化为rna。例如,但不限于,可以使用逆转录酶将mrna生成为互补dna(cdna),并且可以使用rna聚合酶将dna生成为rna。核酸分子可以是生物或合成来源的。核酸分子的实例包括基因组dna、cdna、rna、dna/rna杂交体、扩增的dna、预先存在的核酸文库等。核酸可以从人类样品中获得,例如血液、血清、血浆、脑脊液、脸颊刮屑、活检、精液、尿液、粪便、唾液、汗液等。核酸分子可以进行各种处理,例如修复处理和片段化处理。片段化处理包括机械、声波和流体动力学剪切。修复处理包括缺口修复通过延伸和/或连接、抛光以产生平末端、去除受损碱基,例如脱氨基的、衍生的、脱碱基的或交联的核苷酸等。目标核酸分子也可以进行化学修饰(例如,亚硫酸氢盐转化、甲基化/去甲基化)、延伸、扩增(例如,pcr、等温等)等。
[0140]“互补的”核酸或“互补序列”是能够根据标准的watson
‑
crick、hoogsteen或反向hoogsteen结合互补规则进行碱基配对的核酸。如本文所用,术语“互补的”或“互补序列”可以指基本上互补的核酸,如可以通过上述相同的核苷酸比较所评估的。术语“基本上互补的”可以指包含至少一个序列的连续核碱基或半连续核碱基(如果分子中不存在一个或多个核碱基部分)的核酸能够与至少一条核酸链或双链体杂交,即使少于所有不与对应的核碱基进行碱基配对的核碱基。在某些实施例中,“基本上互补的”核酸含有至少一个序列,其中约70%、约71%、约72%、约73%、约74%、约75%、约76%、约77%、约77%、约78%、约79%、约80%、约81%、约82%、约83%、约84%、约85%、约86%、约87%、约88%、约89%、约
90%、约91%、约92%、约93%、约94%、约95%、约96%、约97%、约98%、约99%、约100%以及其中的任何范围的核碱基序列能够在杂交期间与至少一个单链或双链核酸分子进行碱基配对。在某些实施例中,术语“基本上互补的”是指可以在严格条件下与至少一个核酸链或双链体杂交的至少一个核酸。在某些实施例中,“部分地互补的”核酸包含可以在低严格条件下与至少一个单链或双链核酸杂交的至少一个序列,或包含至少一个序列,其中少于约70%的核碱基序列能够在杂交期间与至少一个单链或双链核酸分子进行碱基配对。
[0141]
术语“非互补的”是指缺乏通过特异性氢键形成至少一个watson
‑
crick碱基对的能力的核酸序列。
[0142]
如本文所用,术语“简并”是指一个核苷酸或一系列核苷酸,其中的同一性可选自多种核苷酸选择,而不是确定的序列。在具体的实施例中,可以选择两种或更多种不同的核苷酸。在进一步的具体实施例中,在一个特定位置处的核苷酸的选择包括选自仅嘌呤、仅嘧啶或选自非配对嘌呤和嘧啶。
[0143]“样品”是指从新鲜或保存的生物样品或合成产生的来源中获得或分离的材料,其中含有目标核酸。样品可包括至少一种细胞、胎儿细胞、细胞培养物、组织标本、血液、血清、血浆、唾液、尿液、泪液、阴道分泌物、汗液、淋巴液、脑脊液、粘膜分泌物、腹膜液、腹水、粪便物质、身体渗出液、脐带血、绒膜绒毛、羊水、胚胎组织、多细胞胚胎、裂解物、提取物、溶液、或怀疑含有目标免疫核酸的反应混合物。样品还可以包括非人类来源,例如非人类灵长类动物、啮齿动物和其他哺乳动物、其他动物、植物、真菌、细菌和病毒。
[0144]
如本文关于核苷酸序列所使用的,“基本上已知”是指具有足够的序列信息以允许制备核酸分子,包括其扩增。尽管在一些实施例中衔接子序列的某些部分是随机的或简并的,但这通常约为100%。因此,在特定实施例中,基本上已知是指约50%至约100%、约60%至约100%、约70%至约100%、约80%至约100%、约90%至约100%、约95%至约100%、约97%至约100%、约98%至约100%或约99%至约100%。
[0145]
x.靶标核酸的进一步加工
[0146]
a.dna的扩增
[0147]
许多模板依赖过程可用于扩增给定模板样品中存在的核酸。最著名的扩增方法之一是聚合酶链式反应(称为pcr
tm
),其详细描述于美国专利号4,683,195、4,683,202和4,800,159以及innis et al.,1990,其中每个均通过引用全部并入本文。简而言之,在存在过量脱氧核苷酸(dntp)和热稳定聚合酶,例如taq(thermus aquaticus)dna聚合酶的情况下,将与待扩增的模板dna的两个区域(每条链一个)互补的两个合成寡核苷酸引物添加到模板dna(不需要是纯的)中。在一系列(通常30
‑
35次)温度循环中,靶标dna反复变性(约90℃),与引物退火(通常在50
‑
60℃下),并从引物延伸出子链(72℃)。当子链被创建,它们在随后的循环中充当模板。因此,两个引物之间的模板区域呈指数扩增,而不是线性扩增。
[0148]
b.dna测序
[0149]
还提供了用于衔接子联接的片段文库的测序的方法。本领域技术人员已知的用于测序核酸的任何技术都可以用于本公开的方法中。dna测序技术包括经典的双脱氧测序反应(sanger方法),该反应使用标记的终止子或引物和在平板或毛细管中进行的凝胶分离;使用可逆终止的标记核苷酸进行边合成边测序;焦磷酸测序;454测序;与标记寡核苷酸文库探针的等位基因特异性杂交;使用与标记克隆文库进行等位基因特异性杂交的边合成边
测序,然后进行连接;在聚合步骤中实时监测标记核苷酸的掺入;以及solid测序。
[0150]
可以使用与illumina测序兼容的方法(例如nextera
tm dna样品制备试剂盒)来生成核酸文库,以及例如在oyola等人(2012)中描述的用于生成illumina下一代测序文库制备的其他方法。在其他实施例中,用与solid
tm
或ion torrent测序方法兼容的方法(例如,fragment文库构建试剂盒、mate
‑
paired文库构建试剂盒、chip
‑
seq试剂盒、total rna
‑
seq试剂盒、sage
tm
试剂盒、rna
‑
seq文库构建试剂盒等)生成核酸文库。例如在pareek(2011)和thudi(2012)中描述的用于下一代测序方法的其他方法,包括可以与本发明的实施例一起使用的用于文库构建的各种方法。
[0151]
在特定方面,在本公开的方法中使用的测序技术包括来自illumina,inc.的hiseq
tm
系统(例如,hiseq
tm 2000和hiseq
tm 1000)nextseq
tm 500系统和miseq
tm
系统。hiseq
tm
系统基于对数百万个片段的大规模并行测序,其使用随机片段化的基因组dna到平面的连接,光学透明的表面和固相扩增,以创建具有数百万个簇的高密度测序流通池,每个簇每平方厘米含有约1,000份模板。这些模板使用四色dna边合成边测序技术进行测序。miseq
tm
系统使用truseq
tm
,illumina基于可逆终止子的边合成边测序。
[0152]
可以在本公开的方法中使用的dna测序技术的另一个实例是454测序(roche)(margulies et al.,2005)。454测序涉及两个步骤。第一步,将dna剪切成大约300
‑
800个碱基对的片段,然后将片段平端化。然后将寡核苷酸衔接子连接至片段的末端。衔接子用作片段的扩增和测序的引物。可以使用例如含有5
′‑
生物素标签的衔接子b将片段连接到dna捕获珠,例如链霉亲和素包被的珠。在油水乳液的液滴内,pcr扩增了与珠连接的片段。结果是每个珠上克隆扩增的dna片段有多个拷贝。在第二步中,将珠捕获在孔(皮升大小)中。对每个dna片段并行进行焦磷酸测序。一个或多个核苷酸的添加产生光信号,所述光信号由ccd相机在测序仪器中记录。信号强度与掺入的核苷酸数量成比例。
[0153]
可以在本公开的方法中使用的dna测序技术的另一个实例是solid技术(life technologies,inc.)。在solid测序中,将基因组dna剪切成片段,并将衔接子连接到片段的5
′
末端和3
′
末端以生成片段文库。可替代地,可以通过将衔接子连接至片段的5
′
末端和3
′
末端、环化片段、消化环化的片段以产生内部衔接子,并将衔接子连接至所得片段的5
′
末端和3
′
末端以生成mate
‑
paired文库来引入内部衔接子。接下来,在含有珠、引物、模板和pcr组分的微反应器中制备克隆珠群体。pcr之后,使模板变性并富集珠以分离具有延伸的模板的珠。对所选珠上的模板进行3
′
修饰,使其可以结合到载玻片上。
[0154]
可以在本公开的方法中使用的dna测序技术的另一个实例是iontorrent系统(life technologies,inc.)。ion torrent使用高密度的微型加工孔阵列以大规模并行方式执行该生化过程。每个孔容纳不同的dna模板。孔下面是离子敏感层,所述离子敏感层下方是专有的离子传感器。如果将核苷酸(例如c)添加到dna模板中,然后掺入dna链中,则会释放出氢离子。来自该离子的电荷会改变溶液的ph值,这可以通过专有的离子传感器进行检测。测序仪将调用碱基,直接从化学信息转变为数字信息。然后,ion personal genome machine(pgm
tm
)测序仪依次用一个接一个的核苷酸淹没芯片。如果淹没芯片的下一个核苷酸不匹配,则不会记录电压变化,也不会调用碱基。如果dna链上有两个相同的碱基,则电压将加倍,并且芯片将记录两个被调用的相同的碱基。因为这是直接检测,没有扫描、没有照
相机、也没有光线,所以每个核苷酸的掺入都以秒为单位记录。
[0155]
可以在本公开的方法中使用的测序技术的另一个实例包括pacific biosciences的单分子实时(smrt
tm
)技术。在smrt
tm
中,四个dna碱基中的每一个都与四种不同荧光染料之一连接。这些染料是磷酸连接的。单个dna聚合酶使用在零模式波导(zmw)底部的单分子模板单链dna固定。zmw是一种限制结构,该限制结构使得能够观察到在快速扩散进zmw和从zmw扩散出来的荧光核苷酸的背景下,dna聚合酶掺入单个核苷酸的情况(以微秒为单位)。将核苷酸掺入生长链中需要花费几毫秒的时间。在这段时间内,荧光标记被激发并产生荧光信号,并且荧光标签被切割掉。染料的相应荧光的检测表明掺入了哪个碱基。重复该过程。
[0156]
另一个测序平台包括cga平台(完整基因组学)。cga技术基于环状dna文库的制备和滚环扩增(rca)以产生排列在固体支持物上的dna纳米球(drmanac等人2009)。完整基因组学的cga平台使用一种称为组合探针锚定连接(cpal)的新型策略进行测序。该过程开始于锚分子和唯一的衔接子之一之间的杂交。四个简并的9
‑
mer寡核苷酸使用特定的荧光团标记,这些荧光团对应于探针第一位置的特定核苷酸(a、c、g或t)。序列测定发生在反应中,其中正确的匹配探针与模板杂交,并使用t4dna连接酶与锚连接。在对连接产物进行成像之后,将连接的锚定探针分子变性。使用新的荧光标记的9
‑
mer探针组将杂交、连接、成像和变性过程重复五次,所述探针在n 1、n 2、n 3和n 4位置含有已知碱基。
[0157]
xi.试剂盒
[0158]
本文的技术包括用于分析dna样品中拷贝数变异或等位基因频率的试剂盒。“试剂盒”是指物理元件的组合。例如,试剂盒可以包括例如一种或多种组分,诸如核酸引物、酶、反应缓冲液、说明书,以及可用于实施本文所述技术的其他元件。可以以适合于执行本发明的任何方式来布置这些物理元件。
[0159]
试剂盒的组分可以包装在以水性介质中或以冻干形式包装。试剂盒的容器器件通常将包括至少一个小瓶、试管、烧瓶、瓶子、注射器或其他容器器件,可以将组分放置在其中,并且优选适当地等分放置(例如,等分到微量滴定板的孔中)。如果试剂盒中有多个组分,则试剂盒通常还将含有第二、第三或其他附加容器,可以将附加组分单独放入其中。但是,组分的各种组合可以包含在单个小瓶中。本发明的试剂盒通常还将包括用于容纳核酸的器件,以及密闭地用于商业销售的任何其他试剂容器。这样的容器可以包括将期望的小瓶保留在其中的注射或吹塑的塑料容器。试剂盒还将包括使用试剂盒组分及试剂盒中未包含的任何其他试剂的使用的说明。说明可以包括可以实现的变体。
[0160]
xii.实例
[0161]
包括以下实施例以说明本发明的优选实施方案。本领域技术人员应该理解,以下实施例中公开的技术代表本发明人发现的在本发明的实践中发挥良好作用的技术,因此可以被视为构成本发明实践的优选模式。然而,根据本公开,本领域技术人员应当理解,在不脱离本发明的精神和范围的情况下,可以对所公开的特定实施例进行许多改变并且仍可获得相同或相似的结果。
[0162]
实例1
‑
校准结果
[0163]
在正常细胞系gdna样品na18562上进行了erbb2 qaseq组合的示例性校准实验,该样品不应包含erbb2扩增,以分析定量变化性(quantitation variability)和潜在的lod。
工作流程如“qaseq工作流程”章节所述。taq聚合酶用于所有pcr步骤。变性在95℃下进行,退火/延伸在60℃下进行(通用pcr步骤除外,其中退火/延伸在68℃下进行)。因为所有附着有umi的原始分子都需要出现在ngs输出中,所以为每个分子/umi保留了15个读段。对于2500个单倍体基因组拷贝和20个扩增子组合的输入,所需的总读段约为2
×
2500
×
20
×
15=1,500,000。请注意,在此工作流程中,一个dna双链体中的每条链都带有不同的umi,因此2500个单倍体基因组拷贝=5000个分子数=8.3ng gdna。该实验在illumina miseq仪器上进行。
[0164]
使用精确的字符串匹配将ngs读段与扩增子序列比对;不同文库的比对率在50%到70%之间。接下来,分析了umi家族规模和唯一umi数量。对于大多数基因座,umi家族规模的分布在≈20处达到峰值(图5)。去除含有明显pcr错误的umi家族(即在poly(h)umi序列中发现的g碱基)和家族规模<4的umi(图5)。如果umi比对率是完美的,唯一umi数量应该等于样品中的原始分子数量。对于2500个单倍体基因组拷贝(5000个分子)的输入,根据基因座获得了632至3065的唯一umi数量(图6)。
[0165]
为了估计该测定的lod,为四种不同的dna输入制备了文库:75、250、750和2500个单倍体基因组拷贝;每个条件重复五次。如“数据分析工作流程”章节所述,计算样品的cnv比率。使用五次重复的cnv比率(σ
cnv比率
)的标准偏差来评估定量变化性;测定的lod可以估计为3σ
cnv比率
。还进行了模拟以计算理论σ
cnv比率
;请注意,如果输入分子数量增加,σ
cnv比率
和lod应减少。σ
cnv比率
高于理论值(图7),这正如预期的那样,因为无法消除umi连接偏差和扩增偏差。对于2500个单倍体基因组拷贝输入,当前最佳σ
cnv比率
为1%;为保守起见,使用了基于所有4个数据点的线性近似,并获得了σ
cnv比率
=2%;因此,估计的lod约为额外拷贝的6%。基于对50,000个单倍体基因组拷贝输入的外推,潜在的σ
cnv比率
为0.3%,且lod约为1%。另一种评估lod的方法是测试一系列包含不同频率的额外拷贝的校准样品;额外拷贝的最低可检测频率是lod。
[0166]
实例2
‑
ffpe样品中的cnv检测结果
[0167]
使用“多重pcr组合设计”章节和实例1中描述的erbb2组合实例分析了两个ffpe载玻片。ffpe载玻片(从asterand购买)来自同一肺癌肿瘤,预期不含erbb2 cnv。首先,使用qiaamp dna ffpe tissue kit(qiagen)提取dna,每个样品获得>6μg的dna。使用与实例1中所述相同的方法制备文库。每个文库使用8.3ng提取的dna,相当于2500个单倍体基因组拷贝和5000个分子输入。为每个文库保留的ngs读段数量(1,500,000个读段)与2500个单倍体基因组拷贝数输入细胞系gdna文库相同。
[0168]
使用与实例1中所述相同的方法进行数据分析。获得了与细胞系gdna文库类似的umi家族规模分布模式(图8a)。唯一umi数量小于具有2500个单倍体基因组拷贝输入细胞系gdna文库。ffpe样品的umi连接率平均约为细胞系gdna的1/4,这表明需要加载300%以上的ffpe dna才能达到与细胞系gdna样品相同的lod(图8b)。
[0169]
ffpe样品的计算的cnv比率示于图8c。该测定的推断lod=15%基于750个单倍体基因组拷贝输入细胞系gdna的校准结果,其具有与ffpe文库相似的唯一umi数量。基于目前的结果,在这些ffpe载玻片中未检测到erbb2的cnv。由于lod随着输入分子数的增加而降低,因此基于2500个单倍体基因组拷贝输入细胞系gdna的校准结果,可以实现6%的lod。
[0170]
实例3
‑
掺入临床ffpe样品中的cnv定量结果
[0171]
100
‑
重qaseq组合用于定量乳腺癌ffpe样品中erbb2的倍性。50
‑
重位于erbb2基因区域(引物序列见表3;引物名称中带有“erbb2”),50
‑
重位于17号染色体的短臂作为参考(引物序列见表3;引物名称中带有“ref”)。
[0172]
将两个先前表征的ffpe dna样品(1个“正常”样品和1个“erbb2扩增异常”样品)混合以生成2.5%、5%和10%的erbb2 fec样品。“正常”样品dna提取自ffpe肺癌样品(购自asterand),该样品不应有erbb2扩增(fec=0%);“erbb2扩增异常”样品dna提取自ffpe乳腺癌样品(购自origene),其erbb2 fec为78%。每个文库的样品输入为8.3ng dna(通过qpcr定量)。“正常”样品使用5个单独制备的重复ngs文库进行测试,每个文库的有8.3ng dna输入。实验归一化fec值显示于图13.归一化fec计算如下:
[0173]
归一化fec
样品
=(1 fec
样品
)/(1 fec
正常样品
)
‑1[0174]
fec
正常样品
是5次重复的平均值。cnv组合的lod估计为:
[0175]
fec
lod
=3
×
σ
正常样品
/(1 fec
正常样品
)=0.85%
[0176]
这里,σ
正常样品
是5次重复的标准偏差。在2.5%、5%和10%的erbb2 fec样品中成功检测到cnv,因为它们计算的fec超出了3个标准偏差范围(见图13)。erbb2的实验归一化fec与预期值密切相关。
[0177]
实例4
‑
用于突变和cnv定量的全面组合
[0178]
所提出的方法(qaseq)不仅可用于cnv定量,还可用于ngs纠错和突变定量。在每个qaseq扩增子中,fp的3
′
和rpin的3
′
之间的区域是突变检测区域(mdr);mdr中的任何小变异(包括碱基取代、缺失和小于500bp的插入)都能以0.1%
‑
0.3%的lod检测到。这比用于突变检测的标准非umi ngs方法要好得多,后者的lod≈1%。
[0179]
开发了一个179
‑
重全面组合,并测试了乳腺癌样品中的突变和cnv定量。每重包含3个引物:fp(又名sfp)、rpin(又名srpb)和rpout(又名srpa),如前述章节所述。95个引物组单独用于cnv定量,其中45个用于基因erbb2,50个用于17号染色体短臂作为参考。用于erbb2基因的5个引物组用于cnv和突变定量。另外79个引物组仅用于突变定量。ufp和urp用于通用扩增(序列见表3)。
[0180]
cnv定量的方法与前述章节相同;突变定量的数据处理工作流程总结于图14.在任选的衔接子修整后,将ngs读段与扩增子序列进行比对。在每个基因座,将读段分入umi家族;去除umi序列有错误的umi家族,并且也删除umi家族规模小(≤3)的umi家族。接下来,找到每个umi家族的共有mdr序列,通常是umi家族中出现次数最多的mdr序列。最后一步是将共有序列与野生型mdr序列进行比较,并执行从头突变调用。一个突变的vaf可以计算为:vaf=具有突变的umi家族数量/umi家族总数。
[0181]
基于horizon discovery的multiplex i cfdna reference standard set,测试该179
‑
重组合。测试了wild type cfdna reference standard的三个重复ngs文库和0.3%cfdna参考标准(通过混合0.1%cfdna reference standard和1%cfdna reference standard来创建)的三个重复。每个文库的样品输入为8.3ng dna(通过qpcr定量)。
[0182]
所有文库的总体在靶率均大于50%(即,>50%的ngs读段可以与扩增子比对);转化率(即测序的输入分子的百分比)平均为62%,并且该组合中的97%具有>10%的转化率(参见图15)。umi校正后的错误率在不同的核苷酸位置有所不同;在horizon discovery multiplex i wild type cfdna reference standard的三个复制文库中,最高错误率为
0.23%、0.20%和0.23%,平均错误率为0.006%、0.005%和0.005%(见图16)。使用0.3%cfdna reference standard验证突变定量能力。6个突变的实验vaf与预期的vaf大体一致;差异主要是由于对少量(≤9)突变分子进行抽样的随机性(见图17)。
[0183]
***
[0184]
鉴于本公开,可以在不进行过度实验的情况下进行和执行本文所公开和要求保护的所有方法。尽管已经根据优选实施例描述了本发明的组合物和方法,但是对于本领域技术人员而言将显而易见的是,在不脱离本发明的概念、精神和范围的情况下,可以对本文所述的方法和对本文所述方法的步骤或对本文所述方法的步骤顺序应用改变。更具体地,将显而易见的是,在化学和生理上均相关的某些药剂可以代替本文所述的药剂,同时将获得相同或相似的结果。对于本领域技术人员显而易见的所有此类类似替代和修改都被认为落入由所附权利要求所限定的本发明的精神、范围和概念内。
[0185]
参考文献
[0186]
以下参考文献在一定程度上提供了示例性的过程或其他细节,所述示例性的过程或其他细节是对本文所述的那些过程或细节的补充,所述参考文献以引用方式明确地并入本文。
[0187]
lun et al.,“noninvasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on dna in maternal plasma,”proc.natl.acad.sci.u.s.a.,105:19920
‑
19925,2008.
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。