作为kcnq增强剂的1
‑
((2
‑
(2,2,2
‑
三氟乙氧基)吡啶
‑4‑
基)甲基)脲衍生物
1.本发明属于医药领域。特别地,本发明涉及用于治疗由运动神经元兴奋性变化引起的肌萎缩性侧索硬化(als)和其它神经系统疾病的化合物、方法和药物组合物,所述疾病包括但不限于原发性侧索硬化、假性延髓麻痹、进行性延髓麻痹、进行性肌肉萎缩和癫痫。
2.als(有时称为“葛雷克氏症(lou gehrig’s disease)”)是一种致命的神经系统疾病,每年影响大约100,000人中的1.5
‑
3人。它的特征是运动神经元逐渐丧失,通常会在诊断后2
‑
3年内导致死亡。尽管研究仍在继续,但大多数als病例似乎是不规则的(sporadic),没有已知原因。
3.als患者通常表现为周围和中枢运动神经元的兴奋性增加,导致肌束震颤、肌肉痉挛和痉挛状态。据认为,增加的神经元兴奋性导致钙超载和细胞死亡。事实上,运动神经元兴奋性与als患者的生存率呈负相关(参见k.kanai等人,j.neurol.neurosurg.psychiatry,83,734
‑
738(2012))。
4.研究调查了als患者中的运动神经元兴奋性变化的机制和原因(与非als患者相比)。(参见k.kanai等人,brain,129,953
‑
962(2006))。研究结果表明神经元中的钾通道活性降低是疾病相关的过度兴奋的主要因素(参见同上)。此外,来自als患者的多能干细胞的运动神经元也表现出过度兴奋。(参见b.j.wainger等人,cell rep.,7,1
‑
11(2014))。这些干细胞衍生的神经元显示延迟整流钾电流降低,并且事实上,当将药剂瑞替加滨和氟吡汀(其是已知的kv7钾通道增强剂)添加到这些干细胞衍生的的神经元中时,运动神经元的过度兴奋性恢复正常并且细胞的体外存活率增加(参见同上)。在另一项研究中,kv7增强剂瑞替加滨在使用大鼠舌下运动神经元的als体外模型中减轻了兴奋毒性症状并增加了存活率(参见f.ghezzi等人,j.physiol.,596,2611
‑
2629(2018))。
5.目前,唯一被批准用于治疗als的药物是利鲁唑,它已经被证明可以将生存期延长2
‑
3个月。显然需要更有效、更好和更持久的治疗。
6.已知的kv7增强剂瑞替加滨作用于kv7.2
‑
7.5(kcnq2
‑
kcnq5)钾通道。它被批准作为耐药性癫痫患者的辅助治疗。它于2011年在欧洲和美国获批,但是2017年被主动撤回。瑞替加滨从临床使用中撤回被认为是基于许多耐受性问题,导致该药物的使用非常有限。耐受性问题包括常见的和可能基于机制的困倦和头晕的发生,以及不太常见的尿潴留和视网膜和皮肤中的色素沉着变化的发生。导致瑞替加滨标签上的黑框警告的视网膜变化和视力丧失的可能性被认为不是基于机制的。尿潴留很可能是膀胱kv7.3/7.5通道增强的结果。然而,鉴于其副作用,需要开发新的kv7增强剂。
7.最近的研究比较了利鲁唑(例如,批准的als治疗方法)和瑞替加滨对als患者运动神经元兴奋性的作用,表明kv7通道的增强剂可能具有优于利鲁唑的功效(参见m.kovalchuk等人,clinical pharmacology&therapeutics,104,1136
‑
1145(2018))。因此,具有比瑞替加滨更好的耐受性的改良增强剂将有益于als和其它过度兴奋相关的障碍的临床治疗。(参见b.kalappa等人,the journal of neuroscience,35(23):8829
–
8842)(2015)。迄今为止,尚未批准作用于kv7的药剂用于治疗als,因此,仍然需要作用于kv7的药
剂,例如提供治疗益处的,替代的kv7增强剂。此外,相对于其它kv7通道,让此类增强剂对kv7.2/7.3更具选择性可能是有益的。还需要一种新的kv7药剂,它可以避免不希望的副作用并且可以提供改善的药理学性质的组合,包括安全性、效力、功效和耐受性,特别是用于治疗周围和中枢运动神经元的兴奋性。
8.本实施方案提供了钾通道增强剂(例如kv7增强剂——也称为kcnq增强剂)的化合物,其可用于治疗由运动神经元兴奋性变化引起的als和其它的神经系统疾病。
9.具体而言,下式化合物(其被标示为“式i”)可以用作kv7增强剂:
[0010][0011]
其中r1是
[0012][0013]
其中,在式i中,r2为h或oh。除了式i的化合物外,一种或多种药学上可接受的盐可以由式i的化合物制成,并且这些药学上可接受的盐也可以被制成并用作kv7增强剂。
[0014]
在一些实施方案中,制备式i的化合物或其药学上可接受的盐使得r1是
[0015][0016]
并且r2是h。
[0017]
在其它的实施方案中,制备式i的化合物(或其药学上可接受的盐)使得r1是
[0018][0019]
并且r2是oh。此外,在此类实施方案中,当r2是oh时,oh基团所连接的碳是立体中心。因此,在一些实施方案中,式i的化合物(或药学上可接受的盐)可以是两种对映异构体的外消旋混合物。在其它的实施方案中,可以使用具体的对映异构体,包括以下对映异构体之一:
[0020][0021][0022]
本领域技术人员将理解如何构建仅使用一种前述对映体的实施方案。其它的实施方案可以设计为具有不同对映异构体的混合物,所述对映异构体对于每一组分具有不同的百分比。
[0023]
在其它的实施方案中,制备式i的化合物(或其药学上可接受的盐)使得
[0024]
r1是
[0025][0026]
并且r2是h。
[0027]
在另外的实施方案中,制备式i的化合物(或其药学上可接受的盐)使得
[0028]
r1是
[0029][0030]
并且r2是oh。此外,在此类实施方案中,当r2是oh时,oh基团所连接的碳是立体中心。因此,在一些实施方案中,式i的化合物(或药学上可接受的盐)可以是两种对映异构体的外消旋混合物。在其它的实施方案中,可以使用具体的对映异构体,包括以下对映异构体之一:
syndromes”rev neurosci.2015;26(2):239
‑
51中找到。因此,本文的化合物和药学上可接受的盐可以用于治疗一种或多种这些疾病。
[0038]
如本文互换使用的,术语“患者”、“受试者”和“个体”是指人,更具体地,指有需要的患者。在某些实施方案中,所述患者的特征还在于患有将受益于kv7的增强的疾病、障碍或病症(例如,als或另一种疾病)。在另一个实施方案中,所述患者的特征还在于处于发展上述病症或发展将受益于kv7的增强的病症的风险中。
[0039]
有效量可以由本领域技术人员通过使用已知技术并通过观察在类似情况下获得的结果来确定。在确定用于患者的有效量时,要考虑许多因素,包括但不限于:其体重(size)、年龄和总体健康;所涉及的特定疾病或障碍;疾病或障碍的程度或涉及或严重程度;个体患者的响应;施用的具体化合物;施用模式;所施用制剂的生物利用度特性;选择的剂量方案;伴随药物的使用;和其它相关情况。在一些实施方案中,有效量提供周围和中枢运动神经元兴奋性的临床显著降低。
[0040]
本发明的化合物配制成药物组合物,其通过任何能使化合物可生物利用的途径施用。优选地,此类组合物用于口服施用。此类药物组合物及其制备方法是本领域公知的(参见,例如remington:the science and practice of pharmacy,l.v.allen,editor,第22版,pharmaceutical press,2012)。
[0041]
式i的化合物及其药学上可接受的盐特别适用于本发明的治疗方法,其优选某些构型。下表的本发明的化合物描述了此类构型。应当理解,这些优选既适用于治疗方法也适用于本发明的化合物。
[0042]
本实施方案的化合物(其中r2为oh)包括:
[0043]
[0044][0045]
在这些化合物中,其中r2是oh,具有上述“平键”的化合物是外消旋的和/或同时存在两种对映异构体(处于50/50的混合物或一些其它的比例)。上面具有楔形键和虚线键的化合物描述了具体的对映异构体。具有“波浪”键的化合物表明它是单一对映异构体,但这种单一对映异构体的确切构型是未知的,因此,此类对映异构体通过它们的旋光度来区分(例如,它们是否沿( )或(
‑
)方向旋转光线)。
[0046]
本实施方案的化合物还包括以下分子(其中r2为h):
[0047][0048]
具有oh基团作为r2的上述化合物可以是具有处于“向上”构型的oh的那些化合物(如楔形投影所标示)。可以设计其它的实施方案,其中oh基团处于“向下”构型。本领域技术人员将理解如何制备该其它的对映异构体,例如通过使用不同的起始材料和/或使用不同的反应等。此类对映异构体具有以下结构并且是当前要求保护的实施方案的一部分:
[0049][0050]
尽管本发明考虑了所有单独的对映异构体和非对映异构体,以及所述化合物的对映异构体的混合物(包括外消旋物),但特别优选上述化合物及其药学上可接受的盐。
[0051]
本领域普通技术人员可以在合成本发明化合物的任何方便点,通过以下方法分离或拆分单一对映异构体,所述方法如选择性结晶技术、手性色谱法(参见例如,j.jacques等人,"enantiomers,racemates,and resolutions",john wiley and sons,inc.,1981,和e.l.eliel与s.h.wilen,”stereochemistry of organic compounds”,wiley
‑
interscience,1994),或超临界流体色谱法(sfc)(参见例如,t.a.berger;“supercritical fluid chromatography primer,”agilent technologies,july 2015)。
[0052]
本发明的化合物的药学上可接受的盐可以,例如在本领域熟知的标准条件下,通过将适合的本发明的化合物的游离碱与适合的药学上可接受的酸在合适的溶剂中反应形成。参见,例如gould,p.l.,“salt selection for basic drugs,”international journal of pharmaceutics,33:201
‑
217(1986);bastin,r.j.等人“salt selection and optimization procedures for pharmaceutical new chemical entities,”organic process research and development,4:427
‑
435(2000);和berge,s.m.等人,“pharmaceutical salts,”journal of pharmaceutical sciences,66:1
‑
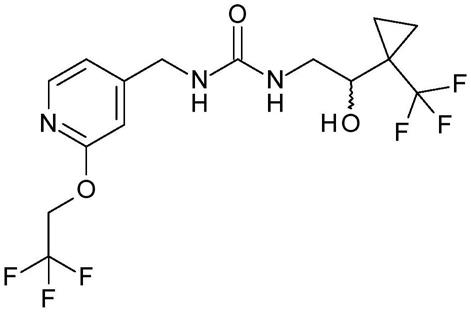
19,(1977)。
[0053]
本发明的化合物或其盐可以通过本领域普通技术人员已知的各种方法制备,其中一些在以下方案、制备和实施例中说明。以下方案中各步骤的产物可以通过本领域熟知的常规方法回收,包括萃取、蒸发、沉淀、层析、过滤、研磨和结晶。在以下方案中,除非另有说明,否则所有取代基如前所定义。试剂和起始材料对于本领域普通技术人员来说是容易获得的。在不限制本发明的范围的情况下,提供以下方案、制备和实施例以进一步说明本发明的方面。此外,本领域普通技术人员理解,式i的化合物可以通过使用可以被本领域技术人员制备的具有相应所需的立体化学构型的起始材料或中间体来制备。
[0054]
下面可能会使用某些缩写。这些缩写的含义如下:“acn”是指乙腈;“ac”是指乙酰基;“acoh”是指乙酸;“ac2o”是指乙酸酐;“ap5”指(2r)
‑
氨基
‑5‑
膦酰基戊酸酯;“bdnf”是指
脑源性神经营养因子;“boc”是指叔丁氧基羰基;“cas#”是指化学文摘登记号;“cmap”是指复合肌肉动作电位;“dcm”是指亚甲基氯或二氯甲烷;“dipea”是指n,n
‑
二异丙基乙胺;“dmea”是指二甲基乙胺;“dmf”是指n,n
‑
二甲基甲酰胺;“dmso”是指二甲亚砜;“d
‑
pbs”是指杜尔贝科的磷酸盐缓冲液;“edta”是指乙二胺四乙酸;“egta”是指乙二醇
‑
双(β
‑
氨基乙基醚)
‑
n,n,n',n'
‑
四乙酸;“es/ms”是指电喷雾质谱;“et2o”是指乙醚;“etoac”是指乙酸乙酯;“etoh”是指乙醇或乙基醇;“h”是指小时(hour/hours);“hepes”是指4
‑
(2
‑
羟基乙基)
‑1‑
哌嗪乙磺酸;“ipa”是指异丙醇或异丙基醇;“ipam”是指异丙胺;“iproac”是指乙酸异丙酯;“lc/msms”是指液相色谱串联质谱;“lihmds”是指双(三甲基甲硅烷基)氨基锂;“kotbu”是指叔丁醇钾;“me”是指甲基;“msec”是指以毫秒(millisecond/milliseconds)作为单位的时间;“mtbe”是指甲基叔丁基醚;“min”是指分钟(minute/minutes);“naotbu”是指叔丁醇钠;“n
‑
buli”是指正丁基锂;“nbqx”是指(2,3
‑
二羟基
‑6‑
硝基
‑7‑
氨磺酰基
‑
苯并[f]喹喔啉;“oac”是指醋酸盐或乙酰氧基;“pbs”是指磷酸盐缓冲盐液;“rt”是指室温;“scx”是指强阳离子交换;“sd”是指标准偏差;“sec”是指以秒(second/seconds)作为单位的时间;“sem”是指平均值的标准误差;“sfc”是指超临界流体色谱法;“tea”指三乙胺;“tfa”指三氟乙酸;“thf”指四氢呋喃;“tma”指三甲胺;“tmeda”指四甲基乙二胺;“tris”指三(羟基甲基)氨基甲烷或2
‑
氨基
‑2‑
(羟基甲基)丙烷
‑
1,3
‑
二醇;“[α]
d20”是指在20℃和589nm的比旋光度,其中c是浓度,单位为g/ml(其通常为g/100ml)。
[0055]
一般方案
[0056]
方案1
[0057][0058]
方案1说明了式i的化合物的制备,其中r1如上文所述定义。适当取代的胺(或其相应的盐,例如hcl)的活化酯形成在本领域中有充分记载,使用,例如羰基二咪唑,和使用(2
‑
乙氧基
‑4‑
吡啶基)甲胺(或其相应的盐形式,例如hcl)与所述活化中间体在具有合适的有机碱的条件下的选择性的n
‑
相对o
‑
羰基化,可以产生本发明所需的脲(步骤1)。本领域技术人员将认识到,含有立体化学的式i的化合物可以通过手性取代的胺制备来获得单一对映异构体,或通过手性拆分式i的化合物,使用手性色谱技术(例如sfc),或通过手性助剂的使用(例如手性盐制剂),这些是本领域所公知。
[0059]
方案2
[0060]
[0061]
方案2描述了外消旋
‑2‑
氨基
‑1‑
[1
‑
(三氟甲基)
‑
环丙基]乙醇盐酸盐的制备。由(三氟甲基)环丙烷甲酸制备weinreb型酰胺(步骤2)可以在本领域中充分描述的各种条件下完成。两步法制备硝基
‑1‑
[1
‑
(三氟甲基)环丙基]乙醇:用标准还原剂还原weinreb型酰胺(步骤3a)(使用,例如在合适的有机溶剂,例如thf或et2o中的氢化铝锂),然后分离醛并将在强碱(如nah或kotbu)存在下生成的硝基甲烷阴离子加成到相应的醛上(步骤3b),可以得到所需的外消旋2
‑
硝基
‑1‑
[1
‑
(三氟甲基)环丙基]乙醇。随后将硝基(步骤4)还原成相应的胺可以在本领域中充分描述的各种条件下完成,并且所得胺可以转化为合适的盐形式,例如hcl,以便于使用。本领域技术人员将认识到,胺的外消旋混合物可以使用本领域熟知的标准技术拆分成其两种手性对映异构体,例如手性色谱法或使用手性盐助剂来制备。
[0062]
方案3
[0063][0064]
方案3描述了必需的(2s)
‑1‑
氨基
‑
3,3
‑
二甲基
‑
丁
‑2‑
醇或其相应的盐的制备。基于jacs第9卷.124,第7期,2002,1307中报道的文献资料,可以使用手性过渡金属催化剂(例如已活化为co
3
的co
2
)对2
‑
叔丁基环氧乙烷进行动力学拆分(步骤5)。所得立体化学可以根据手性催化剂的立体化学进行分配。因此,(2s)
‑2‑
叔丁基环氧乙烷可以使用s,s
‑
(salen)co
3
oac制备(参见jacs第9卷.124,第7期,2002,1307)。此外,可以通过与tetrahedron:asymmetry 13(2002)1209
–
1217中报道的数据进行比较来验证s
‑
立体化学。立体控制的环氧化物开环可以在技术人员熟知的条件下,使用氮亲核试剂,例如在meoh中的nh3来实现(步骤6)。所得氨基醇可以在本领域熟知的条件下转化为合适的盐形式。
[0065]
制备1
[0066]
n
‑
甲氧基
‑
n
‑
甲基
‑1‑
(三氟甲基)环丙烷甲酰胺
[0067][0068]
将市售1
‑
(三氟甲基)环丙烷甲酸[cas#277756
‑
46
‑
4](4.8g,31.4mmol)和n,o
‑
二甲基羟胺盐酸盐(4.65g,46.7mmol)在etoac(50ml)中的混合物冷却至0℃并通过滴液漏斗逐滴加入2,4,6
‑
三丙基
‑
1,3,5,2,4,6
‑
三氧杂三膦烷
‑
2,4,6
‑
三氧化物溶液(50质量%于dmf中,28ml,47.5mmol)。在室温搅拌反应混合物20小时。将反应混合物冷却至0℃,并通过倒入饱和nh4cl水溶液中淬灭。分离所得相并用etoac萃取水层。合并有机萃取液,用mgso4干燥,过滤,并蒸发至干燥,以得到标题化合物。(4.4g,67%产率)。1h nmr(400mhz,cdcl3)δ 3.74(s,3h),3.29(s,3h),1.33
‑
1.25(m,4h).es/ms:m/z198[m h]。
[0069]
制备2
[0070]
(
±
)
‑2‑
硝基
‑1‑
[1
‑
(三氟甲基)环丙基]乙醇
[0071][0072]
将氢化铝锂在thf(20ml,20mmol)中的1m溶液逐滴加入到n
‑
甲氧基
‑
n
‑
甲基
‑1‑
(三氟甲基)环丙烷甲酰胺(4.3g,20.9mmol)在et2o(50ml)中的0℃溶液中,并将所得混合物搅拌1小时。通过逐滴加入水(0.81ml),然后2m naoh水溶液(0.81ml)和额外的水(2.43ml)来淬灭反应。加入mgso4(5g)并在最小真空通过硅藻土垫过滤所得混合物,来得到据信为1
‑
(三氟甲基)环丙烷甲醛在et2o中的溶液。在0℃将此溶液逐滴加入到硝基甲烷(60ml,1.1mol)和kotbu(360mg,3.17mmol)的快速搅拌溶液中。约1小时后,将反应温热至室温并搅拌过夜。从烧瓶中的棕色胶体中滗出溶剂并在减压下浓缩。在硅胶上纯化所得残留物,用0
‑
10%meoh/dcm洗脱,以得到无色油状的标题化合物(2.9g,67%产率)。1h nmr(400mhz,cdcl3)δ 0.97
‑
0.91(m,1h),1.13
‑
0.99(m,3h),2.66(d,j=5.1hz,1h),4.41
‑
4.36(ddd,j=9.8,5.1,2.4hz,1h),4.59
‑
4.53(dd,j=9.8,13.8hz,1h),4.68(dd,j=2.4,13.8hz,1h)。
[0073]
制备3
[0074]
(
±
)
‑2‑
氨基
‑1‑
[1
‑
(三氟甲基)环丙基]乙醇盐酸盐
[0075][0076]
将氢化铝锂在thf(37ml,37mmol)中的1m溶液逐滴加入到(
±
)
‑2‑
硝基
‑1‑
[1
‑
(三氟甲基)环丙基]乙醇(2.9g,14.8mmol)在et2o(30ml)中的0℃溶液里,并搅拌所得反应混合物至室温过夜。将混合物冷却至0℃,并逐滴加入额外的氢化铝锂在thf(15ml,15mmol)中的1m溶液。将反应混合物温热至室温并搅拌4小时。在0℃,通过逐滴加入水(1.96ml),然后2m naoh水溶液(1.96ml),然后水(5.88ml)来淬灭反应混合物。加入mgso4(5g),将所得混合物搅拌10min,并通过硅藻土床过滤。用4m在二噁烷(20ml,80mmol)中的hcl处理滤液并在减压下浓缩。快速搅拌下将所得残留物悬浮在et2o(50ml)中。通过过滤分离所得白色固体,以得到标题化合物(1.52g,47%产率).es/ms:m/z=170[m h]。
[0077]
制备3的替代方法
[0078]
向parr烧瓶中加入氧化铂(iv(242mg,1.07mmol)、( )
‑2‑
硝基
‑1‑
[1
‑
(三氟甲基)环丙基]乙醇(2.42g,11.5mmol)在etoh(24ml)和乙酸(4.96ml)中的溶液。将烧瓶置于55psi的氢气气氛下,并在室温摇动5小时。通过硅藻土床过滤反应混合物并在减压下浓缩。将残余物在1,4
‑
二噁烷(17.2ml)中浆化并逐滴加入在1,4
‑
二噁烷(10ml,40mmol)中的4n hcl并搅拌2小时。过滤混合物并用1,4
‑
二噁烷洗涤滤饼并在真空下干燥15分钟。将所得固体在真空烘箱中于45℃干燥3小时,以得到标题化合物(2.21g,80%产率)。
[0079]
制备4(其得到实施例2)
[0080]
(
±
)
‑1‑
[2
‑
羟基
‑2‑
[1
‑
(三氟甲基)环丙基]乙基]
‑3‑
[[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲基]脲
[0081][0082]
将[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲胺盐酸盐[cas#2044704
‑
69
‑
8](200mg,0.8mmol)在dcm(4ml)中的悬浮液冷却至0℃,加入dipea(580μl,3.3mmol)和1,1
’‑
羰基二咪唑(149mg,0.9mmol)。在0℃剧烈搅拌所得反应混合物15分钟并加入(
±
)
‑2‑
氨基
‑1‑
[1
‑
(三氟甲基)环丙基]乙醇盐酸盐(232mg,1.01mmol)。在室温搅拌所得混合物过夜。通过加入水淬灭,用dcm稀释,并通过疏水性筛板(hydrophobic frit)。蒸发有机层并在硅胶上纯化所得残留物,用0
‑
15%meoh/dcm洗脱,以得到标题化合物(198mg,57%产率)。es/ms:m/z=402[m h]。
[0083]
制备4的替代方法
[0084]
搅拌在dcm(10ml)中的[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲胺二盐酸盐(500mg,1.79mmol)和dipea(1.26ml,7.16mmol),以得到澄清溶液。加入1,1
’‑
羰基二咪唑(311mg,1.88mmol)并搅拌30分钟。加入(
±
)
‑2‑
氨基
‑1‑
[1
‑
(三氟甲基)环丙基]乙醇盐酸盐(465mg,2.15mmol)并在室温搅拌反应混合物48h。加水,分离有机相并用硫酸钠干燥。过滤并蒸发有机物,并在硅胶上纯化所得残留物,用0
‑
15%meoh/dcm洗脱,以得到标题化合物(680mg,90%产率)。
[0085]
该分子的手性部分可以使用动力学拆分和开环化学来合成。
[0086]
制备5
[0087]
(2s)
‑1‑
氨基
‑
3,3
‑
二甲基
‑
丁
‑2‑
醇盐酸盐
[0088][0089]
通过市售获得(2s)
‑2‑
叔丁基环氧乙烷(cas#40102
‑
55
‑
4)或通过j.am.chem.soc.第9卷.124,第7期,2002,1307中报道的动力学拆分获得(2s)
‑2‑
叔丁基环氧乙烷,其中据报道,r构型的环氧化物是由rr催化剂获得的,因此s环氧化物是由ss催化剂获得的:
[0090][0091]
在密封容器中,将(2s)
‑2‑
叔丁基环氧乙烷(107g,1.01moles)和nh4oh(1.3l,10.7moles)在etoh(427ml)中的混合物在100℃加热4小时。冷却并在减压下浓缩。在0℃将所得残留物溶解在dcm(100ml)中,并在10分钟内缓慢加入hcl在二噁烷中的4m溶液(267ml,1.1moles),同时白色沉淀形成。过滤所得固体,用冷dcm洗涤,并通过真空抽吸干燥,以得到标题化合物(103g;62.9%产率)。1h nmr(300.1mhz,meod):d 0.94(s,9h),2.76(dd,j=
11.1,12.6hz,1h),3.09(dd,j=2.5,12.6hz,1h),3.41(dd,j=2.5,11.1hz,1h).[α]
d20
= 32.38
°
(c=0.8g/100ml,etoh)。tetrahedron:asymmetry13(2002)中报道的文献资料1209
–
1217[α]
d20
= 25.9
°
(c=0.47g/100ml,etoh)。
[0092]
制备5的替代方法
[0093]
准备两个标有a和b的isco 2
‑
1000ml注射泵:
[0094]
用7m nh3在meoh中的溶液填充泵a。用溶解在7m nh3在meoh(995ml)中的溶液里的(2s)
‑2‑
叔丁基环氧乙烷(25g,232mmol)溶液填充泵b。将泵连接到烘箱中的500ml不锈钢管反应器(od=1/8”),然后连接到出口,即位于烘箱外的7ml不锈钢管反应器(od=1/16")以作为热交换器,并在热交换器后连接一个设定为1200psi的背压调节器。
[0095]
使用泵a,以5ml/min,用7m nh3在meoh中的溶液填充管式反应器。将烤箱温度设置为200℃。一旦管式反应器填满,切换到泵b以10ml/min运行100min,然后切换到泵a,以相同的流速再输送7m nh3在meoh中的溶液并持续额外的1h。
[0096]
在室温,于减压下浓缩收集的溶液,得到粗制(2s)
‑1‑
氨基
‑
3,3
‑
二甲基
‑
丁
‑2‑
醇(24.4g)。将所述粗制物质溶解在叔丁基甲基醚(150ml)中,并在5分钟内,于剧烈搅拌下逐滴加入5.5m hcl在ipa(46.4ml,255mmol)中的溶液。过滤所得白色固体,用mtbe(4x25ml)洗涤,并干燥以得到标题化合物(23g,72%产率)。
[0097]
制备6
[0098]
[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲胺二盐酸盐
[0099][0100]
将2
‑
(2,2,2
‑
三氟乙氧基)吡啶
‑4‑
甲腈[cas#618446
‑
30
‑
3](82.5g,367mmol)在etoh(500ml)中搅拌,从未溶解的固体中滗出溶液,并且每次滗出溶液用etoh(3x50ml)洗涤固体。合并etoh溶液,用额外的etoh(150ml)稀释,并加入浓hcl水溶液(125ml)。加入10%钯碳(3.7g)在约10ml etoh中的浆液。在室温,于60psi的h2气氛下摇动所得反应混合物过夜。通过硅藻土垫过滤混合物,用etoh洗涤,并在减压下浓缩滤液,以留下白色固体。在45℃将所得固体在mtbe中浆化30min,将混合物冷却至室温,并过滤以得到固体。将固体溶解在水(400ml)中并用mtbe(400然后200ml)萃取两次。在减压下浓缩水相,以得到奶油色固体,将其在thf(100ml)中浆化并过滤。将滤饼用thf(2
×
30ml)洗涤并真空抽吸干燥,以得到标题化合物(46.16g,44%产率)。将滤液进一步在减压下浓缩并在真空烘箱中干燥过夜。将所得固体在thf(50ml)中浆化30min并过滤,以得到另一批标题化合物(34g,32.5%产率)。es/ms:m/z 307[m h]。氯离子分析(ic)显示摩尔比为2:1氯离子:母体。
[0101]
实施例1
[0102]1‑
[(2s)
‑2‑
羟基
‑
3,3
‑
二甲基
‑
丁基]
‑3‑
[[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲基]脲
[0103][0104]
将[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲胺盐酸盐(8.06g,33.2mmol)在dcm(50ml)中搅拌并加入dipea(29ml,166mmol)。将所得混合物搅拌5min并加入1,1'
‑
羰基二咪唑(5.7g,33.2mmol)。将混合物搅拌10min,并加入(2s)
‑1‑
氨基
‑
3,3
‑
二甲基
‑
丁
‑2‑
醇盐酸盐(5g,32.5mmol),并将所得反应混合物搅拌一周末。用水洗涤反应混合物,分离有机相,并在减压下浓缩。通过快速柱色谱纯化所得残留物,用0至10%在dcm中的meoh洗脱,溶剂蒸发后得到标题化合物(8.16g,72%产率)。
[0105]
将材料与另一批如上文所述制备的标题化合物(4.37g)合并并从iproac(45ml)中重结晶,以得到11.29g标题化合物。es/ms:m/z 350[m h]。[α]
d20
= 29.845
°
(c=0.2g/100ml,meoh)。
[0106]
实施例2
[0107]
( )
‑1‑
[2
‑
羟基
‑2‑
[1
‑
(三氟甲基)环丙基]乙基]
‑3‑
[[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲基]脲和(
‑
)1
‑
[2
‑
羟基
‑2‑
[1
‑
(三氟甲基)环丙基]乙基]
‑3‑
[[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲基]脲通过手性拆分
[0108][0109]
使用ad
‑
h(250x30mm,5μ)柱在35℃,100bar对(
±
)
‑1‑
[2
‑
羟基
‑2‑
[1
‑
(三氟甲基)环丙基]乙基]
‑3‑
[[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲基]脲(680mg)进行sfc手性纯化,92:8co2/乙醇和0.2%n,n
‑
二甲基乙胺@152ml/min洗脱并在220nm处检测,蒸发馏分并在45℃真空烘箱中干燥得到:
[0110]
对映异构体1(第一个洗脱峰,285.4mg):(
‑
)
‑1‑
[2
‑
羟基
‑2‑
[1
‑
(三氟甲基)环丙基]乙基]
‑3‑
[[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲基]脲;[α]
d20
=
‑
21.0
°
(c=0.20g/100ml,meoh);
[0111]
对映异构体2(第二个洗脱峰,289.5mg)。使用上述方法对对映异构体2进行第二次sfc纯化;蒸发馏分并在45℃真空烘箱中干燥得到( )
‑1‑
[2
‑
羟基
‑2‑
[1
‑
(三氟甲基)环丙基]乙基]
‑3‑
[[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲基]脲(236mg);[α]
d20
= 15.0
°
(c=0.20g/100ml,meoh)。
[0112]
实施例3
[0113][0114]1‑
[[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲基]
‑3‑
[2
‑
[1
‑
(三氟甲基)环丙基]乙基]脲
[0115]
在圆底烧瓶中,将2
‑
[1
‑
(三氟甲基)环丙基]乙胺盐酸盐(500mg,2.6mmol;参见wo 2013/134252,但也有市售[cas#:1454690
‑
80
‑
2])在dcm(5ml)中搅拌并加入dipea(1.4ml,8mmol)。当得到澄清溶液时,加入1,1'
‑
羰基二咪唑(428mg,2.6mmol)并在室温搅拌所得混合物30min。一次性加入[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲胺二盐酸盐(810mg,2.9mmol),并在室温搅拌30min。将反应混合物温热至40℃并维持3小时,并在室温搅拌16h。将反应混合物转移到微波小瓶中,并在100℃加热30min。通过硅胶色谱纯化所得混合物,用0
‑
10%在dcm中的meoh洗脱,以得到标题化合物(892mg,88%产率)。es/ms:m/z 386[m h]
[0116]
实施例3的替代方法
[0117]
向微波反应容器中加入市售的2
‑
[1
‑
(三氟甲基)环丙基]乙胺盐酸盐(758mg,0.4mmol,cas#561297
‑
93
‑
6),dcm(2ml),dipea(698μl,0.4mmol)和1,1
’‑
羰基二咪唑(649mg,0.4mmol)。在室温摇动5h。在室温,加入0.5m制备好的市售的[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲胺(cas#1454690
‑
80
‑
2;或者,参见wo 2013/134252)在dcm(0.8ml,0.4mmol)中的溶液。在100℃将所得混合物在微波中加热30min。将反应混合物滗入更大的圆底烧瓶中,并用水(5ml)和dcm(5ml)稀释。在室温搅拌15min,并通过疏水性筛板以分离有机相。减压下浓缩滤液并通过高ph反相色谱法纯化所得残留物,其使用75x30mmc18柱,5μ粒径,110a,具有c18 15x30mm保护的axia柱,用23
‑
57%在10mm nh4hco3中的acn(ph~10)洗脱(其中含有5%meoh作为水相),以得到1
‑
[[2
‑
(2,2,2
‑
三氟乙氧基)
‑4‑
吡啶基]甲基]
‑3‑
[2
‑
[1
‑
(三氟甲基)环丙基]乙基]脲(90.6mg,59%产率)。es/ms:m/z 386[m h]
[0118]
测定
[0119]
下面提供了显示本实施方案的化合物在增强kv7方面具有活性的生物测定数据。
[0120]
测定#1
[0121]
在来自als患者ipsc系的运动神经元中,对实施例1的optopatch兴奋性测定
[0122]
皮质神经元和下运动神经元的兴奋性改变是als病理生理学中的一个重要因素
(参见,例如k.kanai等人,j neurol neurosurg psychiatry,83,734
‑
738(2012);p.menon等人,eur j neurol.,24,816
‑
824(2017))。全光学电生理学(“optopatch”)用于评估来自两名具有不同致病突变的als患者的ipsc系的培养的运动神经元的兴奋性。
[0123]
细胞培养:这两个ipsc系分别来自一名具有tardbp致病突变的患者和一名具有c9orf72致病重复扩张突变的患者。通过包含运动神经元形态发生素的双smad抑制神经元模式化方案改编的2d分化方法,从这些系生成运动神经元(参见s.m.chambers等人,nat biotechnol.,27,275
–
280(2009))。通过目视检查、核型分析和对β
‑
iii
‑
微管蛋白和核运动神经元标记isl1染色来验证分化。神经元在mtesr(stem cell technologies)中的单层小鼠胶质上培养,辅以10ng/ml bdnf。成像前48小时,将100nm反式视黄醛(trans
‑
retinal)添加到培养基中。
[0124]
用optopatch载体转导:在体外15天时,将培养的运动神经元用慢病毒载体转导,以驱动执行器cheriff
‑
morange2和电压指示剂quasar3
‑
citrine的共表达(详情参见d.r.hochbaum等人,nat.methods,11,825
–
833(2014))。成像前48小时,将100nm反式视黄醛添加到培养基中。
[0125]
溶液:在含有3mm钾的brainphys
tm
成像缓冲液中进行记录。加入间隙连接阻断剂2
‑
氨基乙氧基二苯基硼酸酯(50μm)以消除细胞之间的电偶联,并使用nbqx(10μm)、gabazine(20μm)和ap5(25μm)来阻止突触传递。
[0126]
optopatch记录:转导五天后,在室温,于定制的超广角(ultra
‑
widefield)荧光显微镜上进行optopatch成像。运动神经元用红色激光激发(200w/cm2;635nm)照射以监测quasar荧光的变化并用蓝色led激发(0
‑
127mw/cm2,470nm)照射,以用cheriff使细胞膜去极化。使用定制的蓝光刺激方案,其由以下组成:i)仅用于监测自发活动的2秒红色照明,ii)增加蓝光强度的5x500毫秒步长和iii)2x2秒线性增加的蓝光渐变(ramps),每个渐变具有不同的最大蓝色强度。optopatch数据是使用hamamatsu orca
‑
flash 4.0scmos相机以1khz帧率记录的。视场大小为4mm x0.5mm。用matlab编写的定制控制软件用于控制照明方案和记录所有影片。为了检查kv7.2/7.3增强剂的急性作用,在成像前将神经元与测试化合物一起孵育15min。
[0127]
数据分析:使用时间主成分分析和空间
‑
时间独立成分分析进行图像分割分析,以隔离单个神经元。使用尖峰寻找算法来寻找动作电位,并通过与载体对照进行比较,分析用于化合物对平均发放频率、适应性、基强度和动作电位波形形状的作用的数据(详情参见c.a.werley等人,curr.protoc.pharmacol.,78,11.20.1
–
11.20.24.(2017))。
[0128]
根据上述方案,实施例1的主要作用是动作电位发放频率对低强度蓝光照明的响应。在5.1mw/cm2的蓝色led照明强度,该化合物以浓度依赖性方式降低动作电位频率。
[0129]
将4参数逻辑方程拟合至数据可以用于确定实施例1对动作电位频率的作用的效能(ec
50
)。结果显示在表1中,分别针对来自携带tardbp和c9orf72突变的患者的系进行的两个单独的分化努力。观察到的效果在性质上相似,但比用已知的kv7.2/7.3增强剂氟吡汀看到的那些效果更有效,证明了实施例1通过降低来自患者的运动神经元的兴奋性来治疗als的潜在效用。
[0130]
表1:抑制来自als患者的运动神经元的兴奋性(以平均值表示(95%置信区间))
[0131][0132]
测定#2
[0133]
kv7增强剂在哺乳动物表达系统中对kv7.2/7.3电导的调节
[0134]
kv7增强剂的效能和功效通过ionworks barracuda(molecular devices)平台上的自动电生理学检查法,使用仪器的群体膜片钳模式进行评估。
[0135]
细胞培养:稳定表达hkv7.2(在四环素诱导下)和hkv7.3(目录号ct6147,charles river)的hek293细胞用于这些研究。细胞维持在杜尔贝科改良的伊格尔培养基/营养混合物ham's f
‑
12(sigma
‑
aldrich)中,辅以5%四环素筛选的胎牛血清(sigma
‑
aldrich)、15mm hepes、500μg/ml g418、100u/ml青霉素、100μg/ml链霉素、29.2mg/ml l
‑
谷氨酰胺、100μg/ml吉欧霉素和5μg/ml杀稻瘟素。hkv7.2的表达是通过在记录前24小时加入1μg/ml多西环素来诱导的。
[0136]
细胞在corning t
‑
150烧瓶中培养至融合度为85%
‑
95%。在实验开始时,将细胞用不含钙和镁的d
‑
pbs洗涤一次,然后在37℃,于3ml 0.25%胰蛋白酶中孵育8分钟以解离。将细胞复悬在培养基中,轻轻研磨并在1,000rpm离心4分钟。细胞在外部溶液中复悬至2.5
‑
3.5m细胞/ml的最终浓度。
[0137]
溶液:外部溶液由以下(以mm计)组成:140nacl、5kcl、2cacl2、1mgcl2、10hepes、10葡萄糖,ph 7.4。内部溶液由以下(以mm计)组成:90k
‑
葡萄糖酸盐、40kcl、3.2egta、3.2mgcl2、5hepes,ph 7.25。膜穿透剂两性霉素b每天制备为27mg/ml的dmso储备溶液,然后添加到内部溶液中至0.1mg/ml的最终浓度。用10mm dmso储备溶液在384孔板中制备测试物品稀释液,并使用声学分配(labcyte)进行稀释,使得最终dmso浓度为0.1%。
[0138]
电生理学记录:将复悬的细胞置于barracuda
tm
(iwb)仪器,将外部溶液添加到384孔膜片板(patch plate)中,并进行孔测试以确定阻断的孔和偏移电压。然后通过仪器将细胞添加到膜片板(每孔9μl)。在将穿透剂两性霉素引入内部溶液之前进行两次密封测试,并允许大约8分钟获得电通路(electrical access),这通过第三次密封测试进行验证。命令电压方案由从
‑
80mv到 40mv的1秒电压阶跃,从
‑
80mv的钳制电位组成,并在添加测试物品之前(基线)和添加6分钟后施加。
[0139]
数据分析:使用iwb软件获取数据并减去泄漏。每个电压阶跃的最后10毫秒期间的电流幅度被平均并输出。使用microsoft excel和graphpad prism进行进一步分析。电流幅度通过以下公式转换为电导(g):g=i/(v
‑
ek),其中i=电流,v=跨步电势,ek=钾反转电位(
‑
84mv)。存在测试物品时的电导被归一化为同一孔在 40mv时的基线电导。电导
‑
电压(g
‑
v)曲线符合玻尔兹曼方程y=bottom (top
‑
bottom)/(1 exp((vm
‑
v0.5/k))。
[0140]
实施例1
‑
3的测试物品介导的电导曲线的中点(v0.5)的偏移显示在表2中。
[0141]
表2:在不同浓度的测试化合物存在下最大半电导中的电压与对照的差异。
[0142][0143][0144]
(上面的sd指的是标准偏差)。
[0145]
将4参数逻辑方程拟合至数据可以用于确定每种测试化合物的效能(ec50)和功效(最大偏移)。实施例1
‑
3的结果示于表3中。
[0146]
表3:kv7.2/7.3增强剂的效能和功效
[0147] 实施例1实施例2实施例3ec
50
(μm)2.01.10.10最大偏移
‑
44
‑
28
‑
23斜率1.21.31.1
[0148]
测定#3
[0149]
阈值追踪以测量ip(在腹腔内)3、10和30mg/kg的实施例1对周围神经兴奋性的作用
[0150]
阈值追踪是一种非侵入性技术,其允许通过提供有关周围轴突的膜电位和离子通道功能的信息来测量其兴奋性性质。
[0151]
方法
[0152]
使用了来自charles river的16只雄性wistar大鼠,重量在307
‑
446g之间。动物在标准饲养条件下分组饲养(每笼4只,07:00h至19:00h光照阶段,恒温(21℃)并恒湿,并且自由获取食物和水,以及环境富集)。
[0153]
用异氟醚(2
‑
2.5%,0.5l/min的o2)麻醉大鼠,然后将其背部放在加热垫上,使尾
部温度保持在32℃以上。环形电极的位置被标记出来,尾巴的标记部分用刀片刮掉以去除毛发和表层皮肤。这些部位用水清洗并干燥,使皮肤和粘性电极之间有良好的传导。
[0154]
将粘性环刺激电极( ve阳极)缠绕在大鼠脚上。将第二个粘性环刺激电极(
‑
ve阴极)缠绕在距尾巴底部1.5厘米处。针记录电极放置在大鼠尾部顶部的中心附近,距离尾部底部的刺激阴极6厘米远。针参比电极放置在大鼠尾部顶部的中心附近,距离记录电极2厘米远。将粘性接地电极缠绕在距记录电极近端2厘米的尾部。
[0155]
研究方案
[0156]
·
启动multitrack程序(如下所述)和spike。
[0157]
·
记录15分钟的稳定基线
[0158]
·
15分钟后,用实施例1或hec给药
[0159]
·
在以下时间点采集血点用于pk:
[0160]
ο给药后10分钟
[0161]
ο给药后20分钟
[0162]
ο给药后30分钟
[0163]
ο给药后40分钟
[0164]
·
ip(腹膜内)给药实施例1后45分钟,给药xe
‑
991(xe
‑
991是一种市售化合物,其在体内福尔马林测定中阻断kcnq增强剂作用——参见y.zheng等人,“activation of peripheral kcnq channels relieves gout pain,”pain,156(2015)1025
–
1035;和r.zaczek,等人,“two new potent neurotransmitter release enhancers,10,10
‑
bis(4
‑
pyridinylmethyl)
‑
9(10h)
‑
anthracenone and 10,10
‑
bis(2
‑
fluoro
‑4‑
pyridinylmethyl)
‑
9(10h)
‑
anthracenone:comparison to linopirdine”the journal of pharmacology and experimental therapeutics,285:724
‑
730,1998。
[0165]
ο(xe
‑
991仅在实施例1之后以30mg/kg给药)
[0166]
·
在以下时间点采集血点用于pk:
[0167]
·
给药后10分钟(实施例1给药后55分钟)
[0168]
·
给药后20分钟(实施例1给药后65分钟)
[0169]
·
给药后30分钟(实施例1给药后75分钟)
[0170]
·
给药后40分钟(实施例1给药后85分钟)
[0171]
在测量大鼠中周围轴突研究的兴奋性性质的阈值追踪测定中,使用1%羟乙基纤维素:0.25%聚山梨醇酯80:0.05%消泡剂:纯净水(hec)制剂以3、10或30mg/kg通过ip给药化合物。在给药后大约10、20、30和最终40分钟收集干血点(dbs)。dbs样品在室温干燥约2小时。在最终时间点获得脑样品并将其冷冻直至分析。dbs样品在室温运输和储存。
[0172]
制备实施例1的1
‑
mg/ml储备溶液并将其连续稀释到合并的大鼠血液中以制备从1至10000ng/ml范围内的标准品。将血液加入到空白dbs卡中以制备标准品。将一个3mm长的dbs标准品或样品加入到96孔板中,然后加入180μl在1:1acn:meoh中的内标(is)。摇动45min,将提取液用水稀释2倍,并通过lc/msms进行药物浓度分析。
[0173]
使用1.14ml的meoh:h2o(2:8)将脑样品均质化。标准品是通过将储备溶液加入到(spiking)一系列5到50000ng/ml范围内的空白脑匀浆中来制备的。将25μl的标准品或样品移入96孔板中,并加入180μl在1:1acn:meoh中的内标(is)并混合。样品在4℃以4000rpm离
心10分钟。上清液用水稀释15倍并通过lc/msms进行分析。
[0174]
样品和标准品使用sciex api 4000三重四极杆质谱仪(sciex,division of mds inc.,toronto,canada)与shimadzu hplc系统(lc
‑
ioad,shimadzu corporation)和gilson 215自动进样器联用进行分析。将样品(0.01ml)注射入5
‑
μm betasil c
‑
18、20x2.1mm javelin(thermo electron corp.cat#70105
‑
022106)的hplc柱,并用梯度洗脱。色谱条件包括流动相a的水/1mnh4hco3(2000:10,v/v)和流动相b的meoh/1m n nh4hco3(2000:10,v/v),其以2.5
‑
min梯度,1.5ml/min的流速运行。带有涡轮喷雾的正离子模式和740℃的离子源温度用于质谱检测。在以下转换中使用多反应监测(mrm)进行定量:在以下转换中使用多反应监测(mrm)进行定量:实施例1(m/z350.2至m/z 233.0)和模拟内标(m/z 263.1至m/z 148.1)。化合物与内标峰面积比相较于药物浓度的线性回归图由1/x2二次方得出。化合物与内标峰面积比相较于药物浓度的线性回归图由1/x2二次方得出。
[0175]
使用的模拟内标是2
‑
(二甲基氨基)
‑
n
‑
戊基
‑3‑
苯基
‑
丙酰胺2,2,2
‑
三氟乙酸(1:1),并且其具有以下结构:
[0176][0177]
该模拟内标购自syncom,其是一家来自荷兰的公司,地址为kadijk 3,9747at groningen,the netherlands。
[0178]
在将药物加入(spiking)到这些基质中并在37℃孵育4.5小时,同时进行轨道振荡后,使用体外透析方法测定药物与大鼠血浆蛋白和脑匀浆的结合。使用ht透析微平衡装置和透析膜带(mwco 12
–
14k)进行测定。在蛋白质基质之后采集时间为0的样品,并在孵育4.5小时后从膜的蛋白质侧和缓冲液侧采集样品。母体在0和45分钟的时间点通过lc
‑
msms进行定量。通过将缓冲液侧的浓度除以蛋白质侧的浓度来计算未结合的分数。还可以通过将缓冲液和蛋白质室的总和除以4.5小时后的时间0浓度来计算回收百分比。未结合的化合物浓度使用总浓度*未结合的分数来计算。
[0179]
结果:实施例1对绝对阈值的作用见表4
[0180]
表4
[0181][0182]
cmap=复合肌肉动作电位
[0183]
(有关该测定的更多信息,请参见r.sittl等人“the kv7 potassiumchannel activator flupirtine affects clinical excitability parameters of myelinated axons in isolated rat sural nerve,”journal of the peripheral nervous system 15:63
–
72(2010);m.kovalchuk等人,“acute effects of riluzole and retigabine on axonal excitability in patients with amyotrophic lateral sclerosis:arandomized,double
‑
blind,placebo
‑
controlled,crossover trial,”2018年3月7日接收;2018年4月13日接受;提前在线出版物00month 2018.doi:10.1002/cpt.1096,clinical pharmacology&therapeutics,第00卷第00号,month 2018;和j.fleckenstein等人“activation of axonal kv7channels in human peripheral nerve by flupirtine but not placebo
‑
therapeutic potential for peripheral neuropathies:results of a randomised controlled trial,”journal oftranslational medicine 2013,11:34.)。
[0184]
时间和时间*治疗相互作用存在显著差异(双向rm anova;时间效应f(26,312)=13.18,p=<0.0001;时间*治疗相互作用f(78,312)=2.888,p<0.0001。bonferroni多重比较测试表明,与媒介物相比,30mg/kg的实施例1显著增加了绝对阈值(兴奋性降低)。xe
‑
991能够逆转这种增加(增加兴奋性)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。