一种no供体化合物及其制备方法和用途
技术领域
1.本发明属于医药技术领域,涉及no供体化合物及其制备方法和用途。
背景技术:
2.在全世界范围内心血管疾病仍然是死亡的主要原因,每年死于心血管疾病的人数高达1730万人,预计到2030年,将有超过2360万的人死于心血管疾病,而缺血性心脏病(ihd)是导致死亡的最主要的原因。
3.硝酸酯类药物是临床上广泛用于治疗ihd的一线药物,主要包括以下四种:硝酸甘油(gtn)、二硝酸异山梨酯(isdn)、5
‑
单硝酸异山梨醇酯(5
‑
ismn)、戊四硝酯(petn)。硝酸酯类药物具有药理活性的机制是它们通过与血管平滑肌和内皮细胞中含巯基的酶相互作用释放出一氧化氮(no),而增加心肌供血和降低心肌耗氧。但是,由于其快速产生耐药性和交叉耐药性,不仅导致治疗效果下降,而且限制了其在临床上提供连续治疗作用。因此,提高硝酸酯类药物的治疗效果,特别是开发无耐药性的硝酸酯类药物已成为心血管领域研究热点。
4.研究表明硝酸酯类药物能够促使血管内产生大量的超氧阴离子自由基而被认为是造成硝酸酯类药物产生耐药性和交叉耐药性的原因之一。因此通过抗氧化剂清除可能是克服硝酸酯类药物耐药性的有效途径。但现有抗氧化剂与硝酸酯类药物联合使用时,存在难以实现药效与药物代谢在同一剂型中协同作用的显著缺陷,导致联合用药受到制约。
5.稳定的氮氧自由基是近年来发展起来的一类新型自由基清除剂,能以“催化”的方式持续分解催化反应的速率常数高达108m
‑1·
s
‑1,是化学计量型自由基清除剂所无法比拟的。而且,某些含有nit型氮氧自由基的化合物具有治疗缺血性心脏病的作用。
6.目前未见到通过引入nit型氮氧自由基形成的双效no供体化合物的报道。
技术实现要素:
7.本发明的目的在于提供一种no供体化合物及其制备方法和用途,该no供体化合物在能够产生no的同时具有较好的抗氧化应激能力,可用于治疗高血压以及缺血性心脏病等心血管疾病。
8.为达到上述目的,本发明采用了以下技术方案:
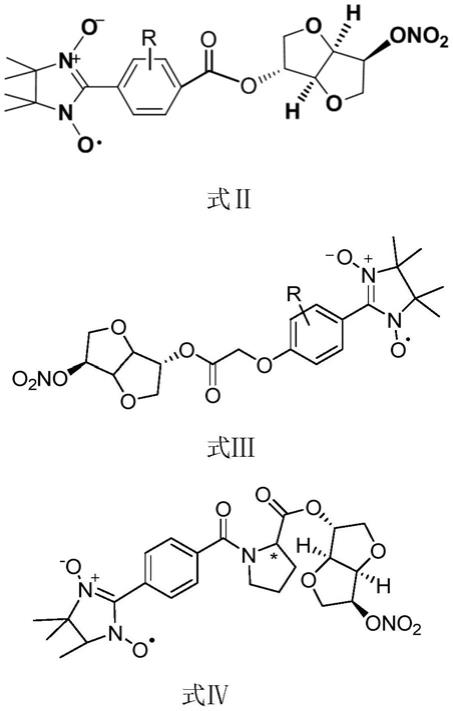
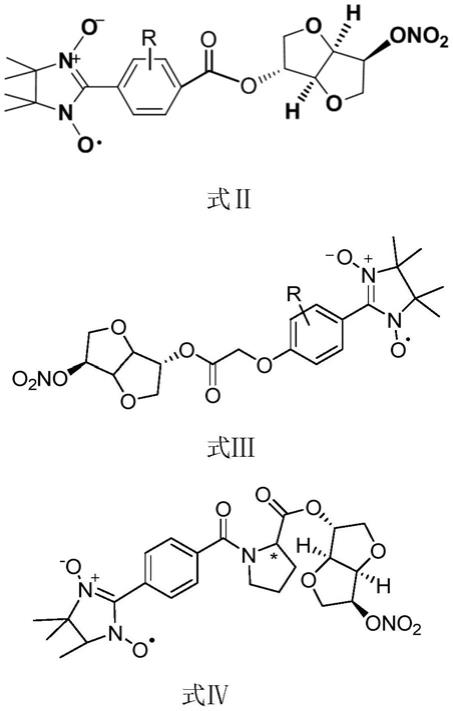
9.一种no供体化合物,该no供体化合物为结构如式i所示的化合物或该化合物的互变异构体、立体异构体、在药学上可接受的盐中的任意一种:
[0010][0011]
其中,连接基团包括用于引入取代基的功能结构,所述功能结构为苯环、取代苯环、乙酸、四氢吡咯环中的任意一种或两种以上的拼接体。
[0012]
优选的,所述连接基团分别与5
‑
单硝酸异山梨醇酯(5
‑
ismn)的羟基部分以及nit型氮氧自由基的2位碳原子相连。
[0013]
优选的,所述no供体化合物为结构如式ⅱ、式ⅲ或式ⅳ所示的化合物或该化合物的互变异构体、立体异构体、在药学上可接受的盐中的任意一种:
[0014][0015]
其中,r为h或取代基,取代基为卤素、卤代烃基、硝基、羟基、烷氧基等各种给电子或吸电子基团中的任意一种(取代基的c原子数不宜过大,否则会导致no供体化合物溶解性不足,例如,烷氧基的c原子数可选为1~3)。
[0016]
优选的,所述no供体化合物为结构如式
ⅱ‑
1至式
ⅱ‑
11所示的化合物或该化合物的互变异构体、立体异构体、在药学上可接受的盐中的任意一种:
[0017][0018]
优选的,所述no供体化合物为结构如式
ⅲ‑
1至式
ⅲ‑
11所示的化合物或该化合物的互变异构体、立体异构体、在药学上可接受的盐中的任意一种:
[0019][0020]
上述no供体化合物的制备方法,包括以下步骤:
[0021]
以5
‑
单硝酸异山梨醇酯(5
‑
ismn)作为形成该no供体化合物结构中的no供体部分的原料,将5
‑
ismn与nit型氮氧自由基通过上述连接基团拼合。
[0022]
优选的,所述no供体化合物的制备方法具体包括以下步骤:
[0023]
2.1)以5
‑
ismn为起始原料,在含有缩合剂、催化剂的溶剂中使5
‑
ismn与对甲酰基苯甲酸或其衍生物进行分子间缩合反应,制得结构如下所示的第一中间体:
[0024]
[0025]
其中,r为h(实施例中化合物1)或取代基,取代基为卤素、卤代烃基、硝基、羟基、烷氧基等各种给电子或吸电子基团中的任意一种,以考察连接基团中引入不同电性取代基团对no供体化合物生物活性的影响;
[0026]
2.2)将第一中间体与双羟胺(指2.3
‑
二甲基
‑
2,3
‑
二羟胺基丁烷)在回流条件下进行缩合反应,制得结构如下所示的第二中间体(例如实施例中化合物2):
[0027][0028]
2.3)将第二中间体与氧化剂进行氧化反应,制得结构如式ⅱ所示的化合物(例如实施例中化合物3)。
[0029]
优选的,所述步骤2.1)中,缩合剂为n,n
′‑
二环己基碳二亚胺(dcc)、羰基二咪唑(cdi)、二异丙基碳二亚胺(dic)、1
‑
(3
‑
二甲胺基丙基)
‑3‑
乙基碳二亚胺(edci)中的一种或多种,催化剂为4
‑
二甲氨基吡啶(dmap)、1
‑
羟基苯并三氮唑(hobt)中的一种或多种;溶剂为四氢呋喃、乙腈、苯、二氯甲烷、n,n
‑
二甲基甲酰胺中的一种或多种;反应温度为25~30℃,反应时间为15~20h,5
‑
ismn与对甲酰基苯甲酸的摩尔比,或者5
‑
ismn与甲酰基苯甲酸衍生物的摩尔比为1:1~1:2,催化剂的用量控制在1%~10%(摩尔比,相对于5
‑
ismn),缩合剂与5
‑
ismn的摩尔比为1:1~1:2;反应结束后过滤,滤液依次经盐酸、饱和碳酸氢钠洗涤后有机相经无水硫酸镁干燥、减压浓缩、柱层析,得到第一中间体。
[0030]
优选的,所述步骤2.2)中,反应所用溶剂为甲醇、乙醇、n,n
‑
二甲基甲酰胺、四氢呋喃中的一种或多种;反应温度为30~80℃,反应时间为12~24h,第一中间体与双羟胺的摩尔比为1:1.1~1:2;反应结束后直接减压浓缩,得到第二中间体(粗品)。
[0031]
优选,所述步骤2.3)中,反应所用溶剂为水、甲醇、四氢呋喃、二氯甲烷或三氯甲烷中的一种或多种,氧化剂为高碘酸钠或二氧化铅中的一种或多种;反应温度为0~25℃,反应时间为30~60min,第一中间体与氧化剂的摩尔比为1:1~1:1.5;反应结束后分离有机相,将有机相依次经饱和氯化钠洗涤、无水硫酸镁干燥、减压浓缩、柱层析,得到no供体化合物。
[0032]
优选的,以上步骤2.1)中,柱层析洗脱剂为乙酸乙酯:石油醚(体积比5:1~1:1),以上步骤2.3)中,柱层析洗脱剂为乙酸乙酯:石油醚(体积比1:5~1:1)。
[0033]
优选的,所述no供体化合物的制备方法具体包括以下步骤:
[0034]
3.1)以对羟基苯甲醛或其衍生物(例如,香草醛等邻位或间位取代的对羟基苯甲醛衍生物)作为起始原料,在碱性条件下与氯乙酸或溴乙酸进行williamson缩合反应,制得结构如下所示的第三中间体:
[0035][0036]
其中,r为h或取代基,取代基为卤素、卤代烃基、硝基、羟基、烷氧基(例如实施例中化合物4)等各种给电子或吸电子基团中的任意一种,以考察连接基团中引入不同电性取代
基团对no供体化合物生物活性的影响;
[0037]
3.2)将第三中间体在含有缩合剂、催化剂的溶剂中与5
‑
ismn进行分子间缩合反应,制得结构如下所示的第四中间体(例如实施例中化合物5):
[0038][0039]
3.3)将第四中间体与双羟胺(指2.3
‑
二甲基
‑
2,3
‑
二羟胺基丁烷)在回流条件下进行缩合反应,制得结构如下所示的第五中间体(例如实施例中化合物6):
[0040][0041]
3.4)将第五中间体与氧化剂进行氧化反应,制得结构如式ⅲ所示的化合物(例如实施例中化合物7)。
[0042]
优选的,所述步骤3.1)中,反应所用的碱为氢氧化钠、氢氧化钾、氢化钠、氢化钾中的一种或多种,反应所用的溶剂为水、四氢呋喃、乙醚中的一种或多种;反应温度为25~80℃,反应时间为5~13h,对羟基苯甲醛(或其衍生物)与氯乙酸的摩尔比,或者对羟基苯甲醛(或其衍生物)与溴乙酸的摩尔比为1:1~1:2,碱的用量控制在2.5~4当量(相对于对羟基苯甲醛或其衍生物的物质的量倍数);反应结束后加水稀释、乙酸乙酯洗涤、分离水相并用浓盐酸酸化、乙酸乙酯萃取、饱和碳酸氢钠分离结合的有机相,水相用浓盐酸酸化、过滤,得到第三中间体。
[0043]
优选的,所述步骤3.2)中,缩合剂为n,n
′‑
二环己基碳二亚胺(dcc)、羰基二咪唑(cdi)、二异丙基碳二亚胺(dic)、1
‑
(3
‑
二甲胺基丙基)
‑3‑
乙基碳二亚胺(edci)中的一种或多种,催化剂为4
‑
二甲氨基吡啶(dmap)、1
‑
羟基苯并三氮唑(hobt)中的一种或多种;溶剂为四氢呋喃、乙腈、苯、二氯甲烷、n,n
‑
二甲基甲酰胺中的一种或多种;反应温度为25~30℃,反应时间为15~36h,5
‑
ismn与第三中间体的摩尔比为1:1~1:2,催化剂的用量控制在1%~10%(摩尔比,相对于5
‑
ismn),5
‑
ismn与缩合剂的摩尔比为1:1~1:2;反应结束后过滤,滤液依次经盐酸、饱和氯化钠洗涤,水相用乙酸乙酯萃取并用无水硫酸镁干燥萃取得到的有机相,减压浓缩、柱层析,得到第四中间体。
[0044]
优选的,所述步骤3.3)中,反应所用溶剂为甲醇、乙醇、n,n
‑
二甲基甲酰胺、四氢呋喃中的一种或多种;反应温度为30~80℃,反应时间为12~24h,第四中间体与双羟胺的摩尔比为1:1.1~1:2,反应结束后直接减压浓缩,得到第五中间体(粗品)。
[0045]
优选的,所述步骤3.4)中,反应所用溶剂为水、甲醇、四氢呋喃、二氯甲烷、三氯甲烷中的一种或多种,氧化剂为高碘酸钠或二氧化铅中的一种或多种;反应温度为0~25℃,反应时间为30~60min,第四中间体与氧化剂的摩尔比为1:1~1:1.5,反应结束后分离有机相,将有机相依次经无水硫酸镁干燥、减压浓缩、柱层析,得到no供体化合物。
[0046]
优选的,以上步骤3.2)、3.4)中,柱层析洗脱剂为乙酸乙酯:石油醚(体积比1:5~
1:1)。
[0047]
优选的,所述no供体化合物的制备方法具体包括以下步骤:
[0048]
4.1)以对甲酰基苯甲酸作为起始原料,与溶剂中含有的酰氯化试剂在回流条件下进行反应,制得第六中间体(实施例中的化合物8);
[0049]
4.2)将第六中间体在含有缚酸剂的溶剂中与l
‑
脯氨酸或d
‑
脯氨酸进行分子间缩合反应,制得第七中间体(例如实施例中化合物9l);
[0050]
4.3)将第七中间体在含有缩合剂、催化剂的溶剂中与5
‑
ismn进行分子间缩合反应,制得第八中间体(例如实施例中化合物10l);
[0051]
4.4)将第八中间体与双羟胺(指2.3
‑
二甲基
‑
2,3
‑
二羟胺基丁烷)在回流条件下进行缩合反应,制得第九中间体(例如实施例中化合物11l);
[0052]
4.5)将第九中间体与氧化剂进行氧化反应,制得结构如式ⅳ所示的化合物(例如实施例中化合物12l)。
[0053]
优选的,所述步骤4.1)中,酰氯化试剂为亚硫酰氯、草酰氯中的一种或多种,溶剂为二氯甲烷、甲苯中的一种或多种;反应温度为80~90℃,反应时间为5~24h,对甲酰基苯甲酸与酰氯化试剂的摩尔比为1:3~1:10;反应结束后减压浓缩,得到第六中间体(粗品)。
[0054]
优选的,所述步骤4.2)中,缚酸剂为三乙胺、二异丙基乙基胺中的一种或多种,溶剂为二氯甲烷、三氯甲烷、甲苯中的一种或多种;反应温度为25~80℃,反应时间为5~20h,对甲酰基苯甲酸与l
‑
脯氨酸的摩尔比,或对甲酰基苯甲酸与d
‑
脯氨酸的摩尔比为1:1~1:2,缚酸剂的用量控制在2.5~4.0当量(相对于对甲酰基苯甲酸的物质的量倍数);反应结束后加水稀释、调节ph至8~10、二氯甲烷洗涤、分离水相并调节ph至2~4、用乙酸乙酯萃取有机相,经无水硫酸钠干燥、减压浓缩、柱层析,得到第七中间体。
[0055]
优选的,所述步骤4.3)中,缩合剂为n,n
′‑
二环己基碳二亚胺(dcc)、羰基二咪唑(cdi)、二异丙基碳二亚胺(dic)、1
‑
(3
‑
二甲胺基丙基)
‑3‑
乙基碳二亚胺(edci)中的一种或多种,催化剂为4
‑
二甲氨基吡啶(dmap)、1
‑
羟基苯并三氮唑(hobt)中的一种或多种,溶剂为四氢呋喃、乙腈、苯、二氯甲烷、n,n
‑
二甲基甲酰胺中的一种或多种;反应温度为25~30℃,反应时间为15~20h,5
‑
ismn与第七中间体的摩尔比为1:1~1:2,催化剂的用量控制在1%~10%(摩尔比,相对于5
‑
ismn),5
‑
ismn与缩合剂的摩尔比为1:1~1:2;反应结束后过滤,滤液依次经盐酸、饱和氯化钠洗涤,水相用乙酸乙酯萃取,萃取得到的有机相用无水硫酸镁干燥,减压浓缩、柱层析,得到第八中间体。
[0056]
优选的,所述步骤4.4)中,反应所用溶剂为甲醇、乙醇、n,n
‑
二甲基甲酰胺、四氢呋喃中的一种或多种;反应温度为30~85℃,反应时间为12~24h,第八中间体与双羟胺的摩尔比为1:1.1~1:2,反应结束后直接减压浓缩,得到第九中间体(粗品)。
[0057]
优选的,所述步骤4.5)中,反应所用溶剂为水、甲醇、四氢呋喃、二氯甲烷、三氯甲烷中的一种或多种,氧化剂为高碘酸钠或二氧化铅中的一种或多种;反应温度为0~25℃,反应时间为30~60min,第八中间体与氧化剂的摩尔比为1:1~1:1.5,反应结束后分离有机相,将有机相依次经无水硫酸镁干燥、减压浓缩、柱层析,得到no供体化合物。
[0058]
优选的,以上步骤4.2)中,柱层析洗脱剂为乙酸乙酯:石油醚(体积比10:1~1:1),以上步骤4.3)、4.5)中,柱层析洗脱剂为乙酸乙酯:石油醚(体积比1:5~1:1)。
[0059]
上述no供体化合物可作为活性成分用于制备治疗缺血性心脏病的药物,尤其可用
于制备治疗心肌缺血/再灌注(mi/r)损伤的药物。
[0060]
优选的,所述no供体化合物具有保护缺血心肌的活性。
[0061]
上述no供体化合物可作为活性成分用于制备治疗高血压的药物。
[0062]
优选的,所述药物的剂型为能够使活性成分有效地到达体内(具体指血液循环系统)的剂型,具体选自片剂、胶囊剂、粉末、颗粒剂、糖浆、溶液、悬浮液、注射剂、酊剂、口服液、气雾剂、口含剂、冲剂、丸剂、散剂等常见剂型或纳米制剂等缓释剂型。
[0063]
优选的,所述药物中,除了含有上述活性成分之外,还可含有少量的且不影响活性成分有效性的次要成分和/或药学上可接受的载体,例如可以含有甜味剂以改善口味、抗氧化剂以防止氧化,以及各种制剂所必要的辅料等。
[0064]
本发明的有益效果体现在:
[0065]
本发明将5
‑
ismn与nit型氮氧自由基拼合,形成了一系列具有抗氧化应激作用的no供体分子(即双功能基团的no供体化合物),可用于制备高血压、缺血性心脏病的治疗药物,该系列的no供体分子中含有氮氧自由基结构单元,解决了抗氧化剂与硝酸酯类药物联合用药存在的难以实现协同作用的问题,有利于拓宽硝酸酯类药物的应用范围和抑制其耐药性。
附图说明
[0066]
图1为由nit型氮氧自由基与5
‑
ismn连接形成的双功能基团的no供体化合物的结构示意图。
[0067]
图2为化合物
ⅱ‑
1的合成路线分步骤示意图之一。
[0068]
图3为化合物
ⅱ‑
1的合成路线分步骤示意图之二。
[0069]
图4为化合物
ⅱ‑
1的单晶结构图。
[0070]
图5为化合物
ⅲ‑
5的合成路线分步骤示意图之一。
[0071]
图6为化合物
ⅲ‑
5的合成路线分步骤示意图之二。
[0072]
图7为化合物
ⅲ‑
5的合成路线分步骤示意图之三。
[0073]
图8为化合物12l的合成路线分步骤示意图之一。
[0074]
图9为化合物12l的合成路线分步骤示意图之二。
[0075]
图10为目标化合物抗氧化损伤细胞活性实验结果(
×
100%)。
[0076]
图11为化合物12l在小鼠高血压模型中的活性实验结果(图中pe表示肾上腺素,ach表示乙酰胆碱,relaxation表示血管舒张程度)。
具体实施方式
[0077]
下面结合附图和实施例对本发明作进一步详细说明。所述实施例仅用于解释本发明,而不用于限制本发明的保护范围。
[0078]
参见图1,本发明将nit型氮氧自由基和5
‑
ismn用连接基团拼合,得到一系列新型no供体化合物,该系列化合物中同时含有氮氧自由基结构单元和5
‑
ismn结构单元,实验结果表明,这样的化合物分子结构对于准确的在no释放位置持续高效地清除具有重要作用,从而在对缺血心肌提供直接的保护作用的同时,还可以克服硝酸酯类药物的耐药性。
[0079]
1.化合物
ⅱ‑
1的制备
[0080]
参见图2,将对甲酰基苯甲酸(0.75g,5mmol)、5
‑
ismn(0.96g,5mmol),及dmap(0.06g,0.5mmol)加入到50ml干燥的四氢呋喃(thf)中,冷却至0℃,加入dcc(1.03g,5mmol),混合物在室温下反应16h,tlc检测原料点消失之后,过滤去除二环己脲,滤液分别用0.5mol/l盐酸洗涤两次,饱和碳酸氢钠水溶液洗涤两次,有机相用无水硫酸镁干燥,真空蒸去溶剂得白色固体,用乙酸乙酯:石油醚=2:1(v/v)的洗脱剂进行柱层析,纯化得到白色固体(化合物1)。化合物1产率66.7%;mp:96.3
‑
98℃;1h nmr(400mhz,cdcl3)δ10.11(s,1h),8.18(d,j=7.2hz,1h),7.96(d,j=7.2hz,1h),5.50(s,1h),5.40(s,1h),5.09(d,j=4.8hz,1h),4.65(d,j=4.8hz,1h),4.23
–
4.15(m,1h),4.15
–
4.01(m,1h),3.95(dd,j=11.2,5.5hz,1h)。
[0081]
参见图3,将化合物1(0.5g,1.55mmol)溶解在15ml甲醇中,加入双羟胺(0.28g,1.86mmol)后升温至80℃回流24h,tlc检测原料消失,真空蒸去溶剂,残渣加入32ml二氯甲烷(dcm)溶解,0℃条件下滴加naio4溶液(naio4为0.33g,1.55mmol),搅拌30min,分离有机层,水相用dcm洗涤,合并有机相后用饱和食盐水洗涤,分离有机层,无水na2so4干燥过滤,真空蒸去溶剂,剩余物用乙酸乙酯:石油醚=1:2(v/v)的洗脱剂进行柱层析,纯化得蓝色固体(化合物3)。化合物3(即化合物
ⅱ‑
1)产率73.1%;ir(kbr,400
‑
4000cm
‑1):3501,2928,2359,1722,1645,1450,1362,1281,1097,852,768,696,542,hrms:450.1499。
[0082]
参见图4,化合物3单晶培养溶剂采用二氯甲烷:乙酸乙酯:石油醚(1:1:5~1:3:5)。单晶结构证明结构式的正确性。
[0083]
2.化合物
ⅲ‑
5的制备
[0084]
参见图5,将香草醛(3g,19.7mmol)溶解在1mol/l naoh中,加入氯乙酸(2.05g,21.7mmol)室温搅拌1h(使酚羟基转变为盐的形式,有利于后续反应)后升温至80℃回流12h,反应结束后,反应液加水稀释,用乙酸乙酯洗涤,分离水相并用浓盐酸(36%~38%)酸化,混合液用乙酸乙酯萃取,加入饱和nahco3水溶液使结合的有机相分离,水相再用浓盐酸酸化,过滤得白色固体(化合物4)。化合物4产率为38%;mp:133.8
‑
135.8℃。
[0085]
参见图6,将化合物4(0.98g,4.66mmol)、5
‑
ismn(0.89g,4.66mmol),及dmap(0.057g,0.47mmol)加入27ml干燥的dcm中,0℃下加入dcc(0.96g,4.66mmol)。混合物在室温下搅拌30h,tlc监测反应完全后,过滤二环己脲沉淀,滤液用0.5mol/l盐酸、饱和nacl水溶液分别洗涤两次,分离有机相,合并有机相用饱和nahco3水溶液洗涤一次,合并水相,水相经乙酸乙酯萃取两次,合并有机相再经无水na2so4干燥后减压旋干,使用乙酸乙酯:石油醚=1:1(v/v)的洗脱剂进行柱层析,纯化得到白色固体(化合物5),化合物5产率为88.97%,mp:122.8
‑
113.6℃;1h nmr(400mhz,cdcl3)δ9.87(s,1h),7.42(dd,j=11.2,3.1hz,2h),6.87(d,j=8.1hz,1h),5.34(dd,j=8.7,2.6hz,2h),4.94(s,1h),4.81(s,2h),4.48(d,j=5.0hz,1h),4.11(d,j=7.2hz,1h),4.07
–
3.97(m,3h),3.95(s,3h),3.90(dd,j=11.4,5.5hz,1h),2.04(s,1h),1.25(d,j=2.0hz,1h)。
[0086]
参见图7,将化合物5(1.48g,3.86mmol)与2.3
‑
二甲基
‑
2,3
‑
二羟胺基丁烷(0.69g,4.63mmol)于30ml甲醇中,升温至80℃回流22h,反应完全后蒸除溶剂,向残渣中加入75ml dcm,冰浴下冷却至0℃后,将naio4(0.83g,3.86mmol)用48ml水溶解后加入其中,搅拌30min,分离有机相,水相用dcm洗涤后合并有机相,有机相经无水naso4干燥后,旋干,使用乙酸乙酯:石油醚=1:2(v/v)的洗脱剂进行柱层析,纯化得深蓝色固体(化合物7)。化合物7
(即化合物
ⅲ‑
5)产率为21.5%;ir(kbr,400
‑
4000cm
‑1):3452,2959,1705,1634,1568,1510,1440,1298,1207,1169,1016,922,912,843,758,735,671,609,515,488.hrms:510.1600。
[0087]
化合物7单晶培养溶剂采用二氯甲烷:乙酸乙酯:石油醚(1:1:5~1:3:5),单晶结构证明结构式的正确性。
[0088]
3.化合物
ⅲ‑
3和
ⅲ‑
8的制备
[0089]
将制备化合物
ⅲ‑
5的起始原料(香草醛)更换为3
‑
溴
‑4‑
羟基苯甲醛和2
‑
溴
‑4‑
羟基苯甲醛,按照相同的方法可以分别制备得到
ⅲ‑
3和
ⅲ‑
8。
[0090]
4.化合物12l的制备
[0091]
参见图8,将对甲酰基苯甲酸(2g,13.3mmol)与二氯亚砜(4.76g,40mmol,即亚硫酰氯)于26ml甲苯中90℃回流5h后,减压蒸去溶剂及过量的亚硫酰氯得淡黄色油状液体,即为对甲酰基苯甲酰氯(化合物8)。在冰浴条件下,将l
‑
脯氨酸(1.53g,13.3mmol)与2ml三乙胺(et3n)溶解在干燥的33ml二氯甲烷中,冰浴下搅拌,将淡黄色油状液体用10ml dcm稀释,然后用恒压漏斗加入反应体系中,室温下反应15h后加水稀释,用1mol/l naoh调节ph至碱性(ph为8~10),经二氯甲烷洗涤后分离水相并调节ph至2~4,用乙酸乙酯多次萃取,合并有机相,用无水na2so4干燥后旋干,用乙酸乙酯:石油醚=10:1(v/v)的洗脱剂进行柱层析,纯化得到2.98g产物(化合物9l)。化合物9l产率90.5%,淡黄色油状物;1h nmr(500mhz,cdcl3)δ10.02(s,1h),8.14(s,2h),7.91(d,j=8.1hz,2h),7.69(d,j=8.1hz,2h),4.67(dd,j=8.4,5.3hz,1h),3.56(dd,j=6.8,3.3hz,1h),3.50
–
3.39(m,1h),2.33(dd,j=13.0,8.0hz,1h),2.18
–
2.07(m,2h),2.01(dd,j=13.1,6.6hz,1h),1.90(dd,j=12.9,6.5hz,1h)。
[0092]
参见图9,化合物9l(2.5g,10.3mmol)、5
‑
ismn(2.0g,10.3mmol)和dmap(0.13g,0.1mmol)溶解在70ml干燥的thf中,0℃下加入dcc(2.83g,13.7mmol),混合物在室温下反应18h,tlc监测反应完全后,过滤沉淀二环己脲,滤液用0.5mol/l盐酸、饱和nacl水溶液分别洗涤两次,分离有机相,有机相用饱和nahco3洗涤一次,合并水相,水相经乙酸乙酯萃取两次,合并有机相再经无水na2so4干燥后,真空除去溶剂,用石油醚:乙酸乙酯=1:1(v/v)的洗脱剂进行柱层析,纯化得到白色固体(化合物10l)。化合物10l产率65%;1h nmr(400mhz,cdcl3)δ9.99(s,1h),7.83(s,2h),7.70(s,2h),4.60(s,1h),4.11(d,j=7.1hz,1h),3.53(dd,j=94.2,54.8hz,4h),2.14(s,1h),2.04(s,1h),1.83(d,j=78.1hz,2h)。
[0093]
将化合物10l(1.76g,4.2mmol)与2.3
‑
二甲基
‑
2,3
‑
二羟胺基丁烷(1.0g,7.14mmol)于80ml甲醇中在85℃条件下回流24h,tlc检测反应完全后,真空旋干,向残渣(油状剩余物)中加入80ml dcm,冰浴下冷却至0℃后,将naio4(0.9g,4.2mmol)用40ml水溶解后加入其中,搅拌30min后分离有机相,水相用dcm洗涤后合并有机相,经无水na2so4干燥后旋干,用石油醚:乙酸乙酯=2:1(v/v)的洗脱剂进行柱层析,纯化得到深蓝色固体(化合物12l)。化合物12l产率83.5%;ir(kbr,400
‑
4000cm
‑1):3445,2955,2923,1745,1632,1414,1389,1364,1306,1281,1202,1167,1132,1082,1020,968,851,766,669,542。
[0094]
化合物12l单晶培养溶剂采用二氯甲烷:乙酸乙酯:石油醚(1:1:5~1:3:5),单晶结构证明结构式的正确性。
[0095]
以上制备的化合物12l的对映异构体为化合物12d,制备工艺除了将l
‑
脯氨酸替换为d
‑
脯氨酸,其他基本相同:
[0096][0097]
5.化合物
ⅴ‑
2的制备
[0098]
在装有回流冷凝管的25ml双颈圆底烧瓶中加入3
‑
溴
‑4‑
羟基苯甲醛(201.02mg,1mmol)、双羟胺(177.85mg,1.2mmol)和8ml甲醇,将混合物在80℃条件下反应60h。反应结束后,将冷却的反应产物转移到50ml圆底烧瓶中,减压除去甲醇,旋干的残渣用8ml二氯甲烷溶解后,在氮气保护作用下置于0℃条件下搅拌,缓慢滴加12.4ml高碘酸钠(213.89mg,1mmol),在0℃条件下反应约30min后分离有机相,水相用dcm洗涤后合并有机相,经无水na2so4干燥后旋干,用石油醚:乙酸乙酯=2:1(v/v)的洗脱剂进行柱层析,纯化得到125.5mg深蓝色固体(化合物
ⅴ‑
2)。化合物
ⅴ‑
2产率38.2%,mp:208.1
‑
210.2℃;ir(kbr,400
‑
4000cm
‑1):2922,1718,1653,1558,1456,1273,1126,1078,964,827;hrms(esi)calculated for c
13
h
16
b
r
n2o3[m h]
:328.04171,found:328.04060;epr(ch2cl2):g factor,2.0083;a
n
,6.5g。
[0099]
将起始原料3
‑
溴
‑4‑
羟基苯甲醛更换为对甲酰基苯甲酸、2
‑
溴
‑4‑
羟基苯甲醛和香草醛,按照相同的方法制备得到
ⅴ‑
1、
ⅴ‑
3和
ⅴ‑
4。制备得到的这些含有nit型氮氧自由基的化合物用于活性对照实验。
[0100][0101]
6.no供体药物片剂的制备实例
[0102]
将化合物
ⅱ‑
1、
ⅲ‑
3、
ⅲ‑
8、
ⅲ‑
5、12l各10g分别与87.5g辅料(白湖精:乳糖=7:3,质量比)混合之后,加入95%的乙醇制粒,干燥,整粒(过筛),加入硬脂酸钠2.5g混合均匀之后压片,得到每片重100mg,化合物
ⅱ‑
1、
ⅲ‑
3、
ⅲ‑
8、
ⅲ‑
5或12l的含量为10mg的片剂。
[0103]
7.no供体药物粉针剂的制备实例
[0104]
将化合物
ⅱ‑
1、
ⅲ‑
3、
ⅲ‑
8、
ⅲ‑
5、12l各1g分别与5g甘露醇溶解于170ml的注射用水中,初次混匀之后,定容到200ml,过滤所得到的溶液,装入西林瓶中,每瓶1ml,冻干,密封、灭菌,得到每支含有5mg化合物
ⅱ‑
1、
ⅲ‑
3、
ⅲ‑
8、
ⅲ‑
5或12l的冻干粉针剂。
[0105]
8.no供体药物胶囊剂的制备实例
[0106]
将化合物
ⅱ‑
1、
ⅲ‑
3、
ⅲ‑
8、
ⅲ‑
5、12l各15g分别与135g辅料(白湖精:乳糖=7:3,质量比)混合之后,加入95%的乙醇制粒,干燥,整粒(过筛),装入胶囊中,每粒重约150mg,其中,化合物
ⅱ‑
1、
ⅲ‑
3、
ⅲ‑
8、
ⅲ‑
5或12l的含量约为15mg。
[0107]
9.内皮细胞的氧化损伤保护(cck
‑
8测定自由基)实验及离体血管舒张功能检测
[0108]
9.1.实验材料
[0109]
①
实验试剂和药品:胎牛血清(fbs),0.25%胰酶
‑
0.02%edta,青霉素
‑
链霉素,低糖dmem基础培养基(ld
‑
dmem),均购自gibco公司;叔丁基过氧化氢溶液(tbhp),cck
‑
8。
[0110]
②
实验细胞:人脐静脉内皮细胞(huvec)
[0111]
9.2.细胞培养
[0112]
配制培养液:取出50ml ld
‑
dmem培养基,然后加入50ml fbs,以及5ml青霉素
‑
链霉素。
[0113]
细胞复苏:将装有huvec细胞的冷冻管在37℃水浴中轻轻震荡溶解,然后将细胞悬浮液缓慢加入15ml的离心管中,在离心机中离心5min,吸出上清液,用配制好的10ml培养液进行重悬,并将重悬液加入10cm细胞培养皿中,放入37℃、5%co2的细胞培养箱中进行培养,并于次日换液。待生长至80%以上时,用0.25%胰酶
‑
0.02%edta溶液消化传代。传代三次后做cck
‑
8实验。
[0114]
9.3双功能基团的no供体化合物对tbhp损伤的huvec的作用
[0115]
取细胞活力良好的对数生长期细胞,用含有胎牛血清的dmem培养液配制成细胞悬液,按每孔6
×
104个/ml的细胞浓度,在96孔板每孔接种100μl,放入37℃、5%co2的细胞培养箱中培养,待细胞过夜贴壁。吸走培养基,空白对照组(ctrl组)加入dmem培养基,给药组、实验组(实验组在给药前预先加入了tbhp)分别加入配制好的含有不同药物(tempol、制备的
ⅱ‑
1、
ⅲ‑
5、
ⅲ‑
3、
ⅲ‑
8、12l等多种双功能基团的no供体化合物以及对照化合物
ⅴ‑
1、
ⅴ‑
2、
ⅴ‑
3、
ⅴ‑
4)的dmem培养基,浓度均为5μmol/ml,tbhp组加入含有tbhp的dmem培养基。每组做4个复孔,每孔各100μl。6h后,吸掉孔板内上清液,在避光的超净工作台内,每孔加入100μl cck
‑
8,并用锡箔纸包裹96孔板,放于37℃的摇床内,低速震荡2h,2h后用酶标仪测定波长450nm处的吸光度值。实验结果表明双功能基团的no供体化合物12l、
ⅲ‑
8、
ⅲ‑
3(图10)对内皮细胞具有较好的保护效应(具有内皮细胞保护效应的化合物还包括
ⅱ‑
1、
ⅲ‑
5、12d),并且
ⅲ‑
8、
ⅲ‑
3等no供体分子可以克服已有no供体分子(例如5
‑
ismn)引起氧化应激的缺点。
[0116]
以上实验结果表明,本发明提出的双功能基团的no供体化合物对人脐静脉内皮细胞(huvec)抗氧化应激模型具有显著的保护作用。因此,可作为活性成分用于制备治疗缺血性心脏病的药物,具有明显的临床应用潜力。
[0117]
为了进一步证实所制备的双功能基团的no供体化合物在治疗缺血性心脏病等心血管疾病中的应用价值,开展了离体血管舒张功能检测。实验结果表明双功能基团的no供体化合物12l、
ⅲ‑
8、
ⅲ‑
3、
ⅱ‑
1、
ⅲ‑
5,及12d具有舒张血管活性。
[0118]
10.双功能基团的no供体化合物药理作用的扩展实验(实验以12l为例)
[0119]
以血管紧张素ii(ang ii)微量泵持续灌注为手段构建小鼠高血压模型,在ang ii灌注的同时给与小鼠腹腔注射12l(20mg/kg,每天注射一次,共28天),在此期间检测小鼠的血压和血管功能。实验结果如图11所示,可以看出12l处理显著降低了高血压模型小鼠的血压,并且改善了高血压模型小鼠的血管功能(图中所示血管舒张程度是实验期间第28天的实验结果)。这些结果不仅验证了以上双功能基团的no供体化合物的舒张血管活性,而且表明其具有用于高血压的治疗的潜能。
[0120]
总之,本发明所述的双功能基团的no供体化合物可以用于制备no供体药物,对高血压、缺血心肌等起到保护作用(且具有开发为无明显耐药性的治疗缺血性心脏病的no供体药物的潜力)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。