一种基于9
‑
芴酮结构骨架的绿光材料及其制备方法和应用
技术领域
1.本发明属于有机绿光发光材料合成领域,涉及一种基于9
‑
芴酮结构骨架的绿光材料及其制备方法和应用。
背景技术:
2.可弯曲的、轻巧的、低成本的、效率高的显示屏一直是人们追求的目标,这样的显示屏将极大地改变人们的日常生活,增进人类的信息互动能力。有机发光二极管(oled)已成为继led之后的下一代显示照明技术,与其他显示技术相比,oled具有视角宽、能耗低、响应速度快、超薄、超轻、成型加工简便等显著优势,可以制备全固化薄膜器件,更可实现柔性显示,得到了人们的广泛关注和深入研究。
3.荧光通常发生在具有刚性平面和π电子共轭体系的分子中,分子内共轭程度的大小是材料荧光性能的主要影响因素,因此,提高分子中π电子共轭程度,可有效提高分子荧光发光效率。芴及芴酮类具有刚性平面联苯结构,分子内含有较大的共轭体系,芴酮类材料具有较高的热稳定性,固体薄膜的荧光量子效果较高,且带隙能较宽,是一类重要的荧光材料。但是因为芴及芴酮本身结构的影响,易形成激基缔合物并且产生长波发射,在器件中会影响整体发射光的色度和颜色的稳定性。因此,提高芴酮类材料的色纯度和稳定性对oled产业发展至关重要。
技术实现要素:
4.为了克服现有技术的不足,本发明的目的之一在于提供一种基于9
‑
芴酮结构骨架的绿光材料,该材料具有发光性能好、热稳定性高、色纯度高的特点。
5.本发明的目的之二在于提供上述基于9
‑
芴酮结构骨架的绿光材料的制备方法。
6.本发明的目的之三在于提供上述基于9
‑
芴酮结构骨架的绿光材料的应用。
7.本发明的目的之一采用如下技术方案实现:
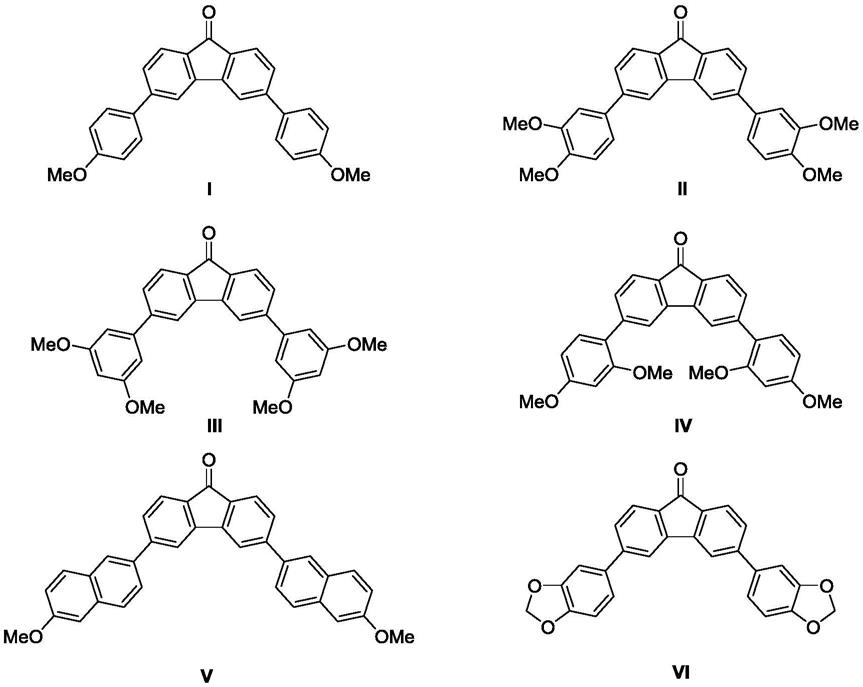
8.一种基于9
‑
芴酮结构骨架的绿光材料,具有结构通式a
[0009][0010]
其中r为r1、r2、r3分别独立选自甲氧基或氢。
[0011]
进一步地,所述的基于9
‑
芴酮结构骨架的绿光材料,其结构为:
[0012][0013]
本发明的目的之二采用如下技术方案实现:
[0014]
基于9
‑
芴酮结构骨架的绿光材料的制备方法,包括以下步骤:
[0015][0016]
在氮气保护下,将3,6
‑
二溴
‑
9h
‑
芴
‑9‑
酮、r对应的硼酸衍生物、四(三苯基膦)钯、碱性物质混合,向其中添加四氢呋喃和水的混合溶液,反应得到最终产物a。
[0017]
进一步地,所述3,6
‑
二溴
‑
9h
‑
芴
‑9‑
酮、r对应的硼酸衍生物、四(三苯基膦)钯、碱性物质的摩尔比例为1:(2~5):(0.006~0.016):(0.8~2)。
[0018]
进一步地,所述r对应的硼酸衍生物选自4
‑
甲氧基苯硼酸、3,4
‑
二甲氧基苯硼酸、3,5
‑
二甲氧基苯硼酸、2,4
‑
二甲氧基苯硼酸、6
‑
甲氧基
‑2‑
萘基硼酸、苯并[d][1,3]二氧杂
‑5‑
基硼酸中的一种。
[0019]
进一步地,所述碱性物质选自碳酸钾、氢氧化钠中的一种。
[0020]
进一步地,所述四氢呋喃、水与3,6
‑
二溴
‑
9h
‑
芴
‑9‑
酮的添加比例为(10~1)ml:1ml:(225.3~22.5)mg。
[0021]
进一步地,所述反应的温度为100℃,时间24~36h。
[0022]
本发明的目的之二采用如下技术方案实现:
[0023]
所述基于9
‑
芴酮结构骨架的绿光材料在有机电致发光器件或智能材料中的应用。
[0024]
相比现有技术,本发明的有益效果在于:
[0025]
本发明提供了一种基于9
‑
芴酮结构骨架的绿光材料,具有发光性能好、热稳定性高、色纯度高的特点。该材料以9
‑
芴酮作为材料母核,通过向9
‑
芴酮的3,6
‑
位引入单取代或多取代的芳基得到绿光材料。借助甲氧基的推电子作用,不同取代位置产生的电子效应和空间效应,使材料载流子的注入和传输更加平衡,有利于提高器件的性能。同时大的位阻作用使得材料具有特定的空间扭转角,有助于提高固态薄膜的荧光量子效率,使得材料可以在有机溶剂中进行高效绿光发射。本发明还提供了上述绿光材料的制备方法,该方法具有原料成本低、反应条件温和、操作简单、产率高等优点。本发明还提供了上述绿光材料在有机电致发光器件或智能材料中的应用,该材料为纯有机小分子绿光材料,能够有效克服现有电致发光材料的热稳定性低及废弃发光器件重金属污染的缺点,具有良好的应用前景。
附图说明
[0026]
图1为本发明实施例1至实施例6的合成路线图;
[0027]
图2为本发明实施例1得到的产物的1h nmr图;
[0028]
图3为本发明实施例1得到的产物的
13
c nmr图;
[0029]
图4为本发明实施例2得到的产物的1h nmr图;
[0030]
图5为本发明实施例2得到的产物的
13
c nmr图;
[0031]
图6为本发明实施例3得到的产物的1h nmr图;
[0032]
图7为本发明实施例3得到的产物的
13
c nmr图;
[0033]
图8为本发明实施例4得到的产物的1h nmr图;
[0034]
图9为本发明实施例4得到的产物的
13
c nmr图;
[0035]
图10为本发明实施例5得到的产物的1h nmr图;
[0036]
图11为本发明实施例5得到的产物的
13
c nmr图;
[0037]
图12为本发明实施例6得到的产物的1h nmr图;
[0038]
图13为本发明实施例6得到的产物的
13
c nmr图;
[0039]
图14为本发明实施例1得到的产物的发射光谱图;
[0040]
图15为本发明实施例2得到的产物的发射光谱图;
[0041]
图16为本发明实施例3得到的产物的发射光谱图;
[0042]
图17为本发明实施例4得到的产物的发射光谱图;
[0043]
图18为本发明实施例5得到的产物的发射光谱图;
[0044]
图19为本发明实施例6得到的产物的发射光谱图;
[0045]
图20为本发明实施例5得到的产物的热重分析图;
[0046]
图21为本发明实施例6得到的产物的热重分析图。
具体实施方式
[0047]
下面,结合附图以及具体实施方式,对本发明做进一步描述,需要说明的是,在不相冲突的前提下,以下描述的各实施例之间或各技术特征之间可以任意组合形成新的实施例。
[0048]
实施例1至实施例6化合物的合成路线如图1所示。
[0049]
实施例1
[0050]
3,6
‑
双(4
‑
甲氧苯基)
‑
9h
‑
芴
‑9‑
酮的合成:
[0051][0052]
将3,6
‑
二溴
‑
9h
‑
芴
‑9‑
酮(67.6mg,0.2mmol)、4
‑
甲氧基苯硼酸(91.2mg,0.6mmol)、四(三苯基膦)钯(11.6mg,0.01mmol)、碳酸钾(165.8mg,1.2mmol)加入到schlenk管中。在氮气保护下,加入3ml四氢呋喃:水=3:1混合溶液,加热至100℃反应36小时。反应结束后,冷却至室温,向反应液中缓慢加入饱和nacl溶液,并用3
×
10ml二氯甲烷萃取。合并有机相,加入无水na2so4干燥,过滤除去na2so4,减压蒸干有机溶剂,粗产品通过硅胶柱层析(石油醚:二氯甲烷=1:5)得到产品3,6
‑
双(4
‑
甲氧苯基)
‑
9h
‑
芴
‑9‑
酮ⅰ,收率85%。3,6
‑
双(4
‑
甲氧苯基)
‑
9h
‑
芴
‑9‑
酮ⅰ的1h nmr图、
13
c nmr图分别如图2、图3所示。
[0053]1h nmr(400mhz,cdcl3)δ7.74(d,j=1.5hz,2h),7.70(d,j=7.7hz,2h),7.66
–
7.58(m,4h),7.48(dd,j=7.7,1.5hz,2h),7.05
–
6.99(m,4h),3.88(s,6h).
13
c nmr(100mhz,cdcl3)δ193.2,160.1,147.3,144.9,133.1,132.6,128.4,127.4,124.7,118.6,114.4,55.4.
[0054]
实施例2
[0055]
3,6
‑
双(3,4
‑
二甲氧苯基)
‑
9h
‑
芴
‑9‑
酮的合成:
[0056][0057]
将3,6
‑
二溴
‑
9h
‑
芴
‑9‑
酮(67.6mg,0.2mmol)、3,4
‑
二甲氧基苯硼酸(109.2mg,0.6mmol)、四(三苯基膦)钯(11.6mg,0.01mmol)、氢氧化钠(48mg,1.2mmol)加入到schlenk管中。在氮气保护下,加入3ml四氢呋喃:水=3:1混合溶液,加热至100℃反应24小时。反应结束后,冷却至室温,向反应液中缓慢加入饱和nacl溶液,并用3
×
10ml二氯甲烷萃取。合并有机相,加入无水na2so4干燥,过滤除去na2so4,减压蒸干有机溶剂,粗产品通过硅胶柱层析(石油醚:二氯甲烷=1:5)得到产品3,6
‑
双(3,4
‑
二甲氧苯基)
‑
9h
‑
芴
‑9‑
酮ⅱ,收率91%。3,6
‑
双(3,4
‑
二甲氧苯基)
‑
9h
‑
芴
‑9‑
酮ⅱ的1h nmr图、
13
c nmr图分别如图4、图5所示。
[0058]1h nmr(400mhz,cdcl3)δ7.75(s,2h),7.72(d,j=7.6hz,2h),7.49(d,j=7.7hz,2h),7.25(d,j=8.8hz,2h),7.17(s,2h),6.99(d,j=8.2hz,2h),4.00(s,6h),3.95(s,6h).
13
c nmr(100mhz,cdcl3)δ193.2,149.7,149.4,147.7,145.0,133.3,133.2,127.7,124.8,120.0,118.9,111.5,110.4,56.24,56.16.
[0059]
实施例3
[0060]
3,6
‑
双(3,5
‑
二甲氧苯基)
‑
9h
‑
芴
‑9‑
酮的合成:
[0061][0062]
将3,6
‑
二溴
‑
9h
‑
芴
‑9‑
酮(67.6mg,0.2mmol)、3,5
‑
二甲氧基苯硼酸(109.2mg,0.6mmol)、四(三苯基膦)钯(11.6mg,0.01mmol)、氢氧化钠(48mg,1.2mmol)加入到schlenk管中。在氮气保护下,加入3ml四氢呋喃:水=3:1混合溶液,加热至100℃反应24小时。反应结束后,冷却至室温,向反应液中缓慢加入饱和nacl溶液,并用3
×
10ml二氯甲烷萃取。合并有机相,加入无水na2so4干燥,过滤除去na2so4,减压蒸干有机溶剂,粗产品通过硅胶柱层析(石油醚:二氯甲烷=1:5)得到产品3,6
‑
双(3,5
‑
二甲氧苯基)
‑
9h
‑
芴
‑9‑
酮ⅲ,收率88%。3,6
‑
双(3,5
‑
二甲氧苯基)
‑
9h
‑
芴
‑9‑
酮ⅲ的1h nmr图、
13
c nmr图分别如图6、图7所示。
[0063]1h nmr(400mhz,cdcl3)δ7.75(s,2h),7.72(d,j=7.6hz,2h),7.51(d,j=7.5hz,2h),6.79(s,4h),6.53(s,2h),3.88(s,12h).
13
c nmr(100mhz,cdcl3)δ193.2,161.3,147.9,144.9,142.5,133.9,128.3,124.8,119.4,105.6,100.4,55.7.
[0064]
实施例4
[0065]
3,6
‑
双(2,4
‑
二甲氧苯基)
‑
9h
‑
芴
‑9‑
酮的合成:
[0066][0067]
将3,6
‑
二溴
‑
9h
‑
芴
‑9‑
酮(67.6mg,0.2mmol)、2,4
‑
二甲氧基苯硼酸(109.2mg,0.6mmol)、四(三苯基膦)钯(11.6mg,0.01mmol)、氢氧化钠(48mg,1.2mmol)加入到schlenk管中。在氮气保护下,加入3ml四氢呋喃:水=3:1混合溶液,加热至100℃反应24小时。反应结束后,冷却至室温,向反应液中缓慢加入饱和nacl溶液,并用3
×
10ml二氯甲烷萃取。合并有机相,加入无水na2so4干燥,过滤除去na2so4,减压蒸干有机溶剂,粗产品通过硅胶柱层析(石油醚:二氯甲烷=1:5)得到产品3,6
‑
双(2,4
‑
二甲氧苯基)
‑
9h
‑
芴
‑9‑
酮ⅳ,收率84%。3,6
‑
双(2,4
‑
二甲氧苯基)
‑
9h
‑
芴
‑9‑
酮ⅳ的1h nmr图、
13
c nmr图分别如图8、图9所示。
[0068]1h nmr(400mhz,cdcl3)δ7.72
–
7.61(m,4h),7.41(d,j=7.6hz,2h),7.31(d,j=8.1hz,2h),6.69
–
6.46(m,4h),3.87(s,6h),3.83(s,6h).
13
c nmr(100mhz,cdcl3)δ193.7,161.2,157.7,145.2,144.5,133.0,131.3,130.1,124.0,122.8,121.7,104.9,99.2,55.8,55.6.
[0069]
实施例5
[0070]
3,6
‑
双(6
‑
甲氧基萘
‑2‑
基)
‑
9h
‑
芴
‑9‑
酮的合成:
[0071][0072]
将3,6
‑
二溴
‑
9h
‑
芴
‑9‑
酮(67.6mg,0.2mmol)、6
‑
甲氧基
‑2‑
萘基硼酸(121.2mg,0.6mmol)、四(三苯基膦)钯(11.6mg,0.01mmol)、氢氧化钠(48mg,1.2mmol)加入到schlenk管中。在氮气保护下,加入3ml四氢呋喃:水=3:1混合溶液,加热至100℃反应24小时。反应结束后,冷却至室温,向反应液中缓慢加入饱和nacl溶液,并用3
×
10ml二氯甲烷萃取。合并有机相,加入无水na2so4干燥,过滤除去na2so4,减压蒸干有机溶剂,粗产品通过硅胶柱层析(石油醚:二氯甲烷=1:5)得到产品3,6
‑
双(6
‑
甲氧基萘
‑2‑
基)
‑
9h
‑
芴
‑9‑
酮
ⅴ
,收率76%。3,6
‑
双(6
‑
甲氧基萘
‑2‑
基)
‑
9h
‑
芴
‑9‑
酮
ⅴ
的1h nmr图、
13
c nmr图分别如图10、图11所示。
[0073]1h nmr(400mhz,cdcl3)δ8.09(s,2h),7.95(s,2h),7.85(t,j=8.0hz,4h),7.79(d,j=8.0hz,4h),7.66(d,j=7.7hz,2h),7.24
–
7.16(m,4h),3.96(s,6h).
13
c nmr(100mhz,cdcl3)δ193.2,158.3,147.8,145.0,135.3,134.4,133.4,129.9,129.0,128.1,127.6,126.2,125.6,124.8,119.6,119.2,105.6,55.4.
[0074]
实施例6
[0075]
3,6
‑
双(苯并[d][1,3]二氧杂
‑5‑
基)
‑
9h
‑
芴
‑9‑
酮的合成:
[0076][0077]
将3,6
‑
二溴
‑
9h
‑
芴
‑9‑
酮(67.6mg,0.2mmol)、苯并[d][1,3]二氧杂
‑5‑
基硼酸(99.5mg,0.6mmol)、四(三苯基膦)钯(11.6mg,0.01mmol)、氢氧化钠(48mg,1.2mmol)加入到schlenk管中。在氮气保护下,加入3ml四氢呋喃:水=3:1混合溶液,加热至100℃反应24小时。反应结束后,冷却至室温,向反应液中缓慢加入饱和nacl溶液,并用3
×
10ml二氯甲烷萃取。合并有机相,加入无水na2so4干燥,过滤除去na2so4,减压蒸干有机溶剂,粗产品通过硅胶柱层析(石油醚:二氯甲烷=1:5)得到产品3,6
‑
双(苯并[d][1,3]二氧杂
‑5‑
基)
‑
9h
‑
芴
‑9‑
酮
ⅵ
,收率92%。3,6
‑
双(苯并[d][1,3]二氧杂
‑5‑
基)
‑
9h
‑
芴
‑9‑
酮
ⅵ
的1h nmr图、
13
c nmr图分别如图12、图13所示。
[0078]1h nmr(400mhz,cdcl3)δ7.71(dd,j=7.3,1.8hz,4h),7.45(d,j=7.7hz,2h),7.19
–
7.10(m,4h),6.93(d,j=7.8hz,2h),6.05(s,4h).
13
c nmr(100mhz,cdcl3)δ193.1,148.5,148.2,147.6,145.0,134.6,133.4,127.8,124.8,121.3,119.0,108.9,107.7,101.6.
[0079]
实验例
[0080]
发射光谱检测:
[0081]
使用实施例1至实施例6制备得到的化合物,用容量瓶配制成10
‑4mol/l的二氯甲烷标准溶液。将实施例1至实施例6制备得到的化合物分别检测发射光谱,结果如图14至19所
示。由图中可以看出实施例1至实施例6制备得到的化合物发光峰波长为510nm左右,对应绿光光区。
[0082]
热重分析:
[0083]
分别称量实施例5、实施例6制备得到的化合物v 3.620mg和化合物vi 3.867mg在sta409pc型热分析仪上进行热重分析。设定最高温度为1050℃,测试温度范围:30℃—1050℃,化合物均在氩气保护下进行测试。结果如图20、图21所示:从室温升温至最终检测温度963.5℃,化合物v和化合物vi分别失重35.12%、23.17%,表明本发明制备得到的化合物在较高温度下具有热稳定性高的特点。
[0084]
本发明提供了一种基于9
‑
芴酮结构骨架的绿光材料,该材料以9
‑
芴酮作为材料母核,通过向9
‑
芴酮的3,6
‑
位引入单取代或多取代的芳基得到绿光材料。借助甲氧基的推电子作用,不同取代位置产生的电子效应和空间效应,使材料载流子的注入和传输更加平衡,有利于提高器件的性能。同时大的位阻作用使得材料具有特定的空间扭转角,有助于提高固态薄膜的荧光量子效率,使得材料可以在有机溶剂中进行高效绿光发射。本发明还提供了上述绿光材料的制备方法,该方法具有原料成本低、反应条件温和、操作简单、产率高等优点。本发明还提供了上述绿光材料在有机电致发光器件或智能材料中的应用,该材料为纯有机小分子绿光材料,具有色纯度高的特点,能够有效克服现有电致发光材料的热稳定性低及废弃发光器件重金属污染的缺点,具有良好的应用前景。
[0085]
上述实施方式仅为本发明的优选实施方式,不能以此来限定本发明保护的范围,本领域的技术人员在本发明的基础上所做的任何非实质性的变化及替换均属于本发明所要求保护的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。