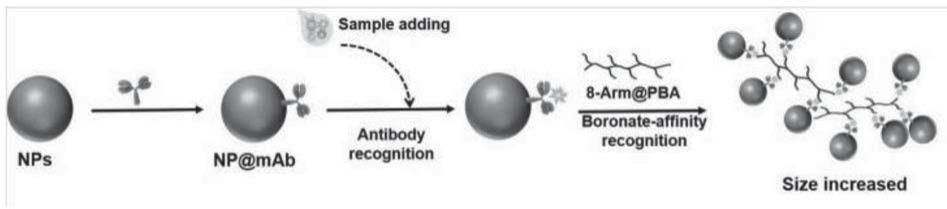
1.本发明涉及免疫分析技术领域,具体为一种基于苯硼酸交联剂的糖蛋白动态光散射免疫方法。
背景技术:
2.近年来,硼酸作为重要的配体被广泛的应用于构建功能化材料,被广泛的应用于传感器中检测顺式二醇的生物分子,硼酸亲和作用的ph开关性质使苯硼酸成为识别和富集含有顺式二醇物质的重要分子,硼酸与顺式二醇结合的独特性质使识别分子苯硼酸可作为抗体替代物用于仿生免疫学分析,因此,可在节约一个抗体的基础上实现对顺式二醇物质(糖蛋白)的检测,从而大大减少了对配对抗体的需求。
3.动态光散射技术,又称光子相关光谱或准弹性光散射,是一种广泛应用于粒度和粒度分布研究的技术,基于动态光散射的均相免疫传感器因其超高的灵敏度及特异性,在各种化学和生物目标检测方面受到了越来越多的关注,另外,磁性纳米材料因其独特的物理和化学性质已成为目前研究应用较多的一类纳米材料,具有良好的分散性及富集能力,生物相容性好、可操控性强等优良性能,因此被广泛应用于免疫学分析、蛋白的分离与纯化等。
4.免疫分析法是应用最广泛的糖蛋白分析方法之一,目前,酶联免疫吸附法(elisa)以其高通量和良好的鲁棒性等独特优势,在致病菌和蛋白生物标志物检测方面占据主导地位,然而,一对完全匹配且对目标物具有高亲和力和特异性的抗体是建立高灵敏和特异性免疫分析方法的必要条件,但获得高度匹配的抗体具有较大的难度,此外,传统的夹心elisa使用酶催化发色底物以产生有色产物作为信号输出,灵敏度通常介于ng/ml至g/ml之间;同时,天然酶贮存稳定性及对环境的耐受性差,因此开发一种灵敏、无酶、无需配对抗体的检测新方法具有重要的意义。
技术实现要素:
5.针对现有技术的不足,本发明提供了一种基于苯硼酸交联剂的糖蛋白动态光散射免疫方法,具备既节约了一个抗体解决配对抗体制备困难的问题,又实现了对糖蛋白的高灵敏及时检测的优点,解决了现有糖蛋白的主要检测方法传统酶联免疫吸附法检测操作步骤复杂、抗原抗体利用和反应效率偏低、灵敏度低、信号不稳定及假阳性信号等的问题。
6.为实现上述目的,本发明提供如下技术方案:一种基于苯硼酸交联剂的糖蛋白动态光散射免疫方法,所述苯硼酸交联剂和免疫识别元件标记的磁性载体作为动态光散射信号增强探针以目标糖蛋白为桥梁形成“多层夹心”结构前后溶液的平均水化动力学粒径变化作为动态光散射信号输出,利用水化动力学直径变化测定反应待检样品中糖蛋白的含量,包括以下步骤:
7.(1)磁性探针:采用edc一步法将糖蛋白特异性免疫识别元件标记羧基化磁性载体表面,获得特异性免疫识别元件标记的磁性探针;
8.(2)硼酸交联剂:通过载体共价偶联苯硼酸形成硼酸交联剂复合物,用于对糖蛋白的识别;
9.(3)糖蛋白的检测:在待测目标物溶液中加入特异性免疫识别元件标记的磁性探针和苯硼酸交联剂,37℃反应15
‑
30min后于马尔文纳米粒度仪上测定溶液的平均水化动力学直径,利用水化动力学直径变化测定反应待检样品中糖蛋白的含量。
10.进一步,所述方法针对目标糖蛋白的特异性免疫识别元件单克隆抗体或者适配体等。
11.进一步,所述磁性载体为fe304纳米微球、金磁颗粒、硅包磁、磁珠等。
12.进一步,所述硼酸交联剂为蛋白、纳米粒子、聚合物作为载体连接苯硼酸形成的复合物等。
13.进一步,所述步骤(1)中采用edc一步法将糖蛋白特异性免疫识别元件标记于磁性载体表面形成磁性免疫探针,所述特异性免疫识别元件标记的羧基化磁性载体的具体制备方法为:取磁性载体于ph6.0
‑
8.0pb(0.01mol/l)缓冲液中,加入特异性免疫识别元件后室温下搅拌反应,1
‑
乙基
‑
(3
‑
二甲基氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edc)重复加3次,反应完成磁吸5
‑
20min后弃上清;随后加入质量体积分数为2%的酸水解酪蛋白,并加入edc,室温搅拌1h后,磁吸5
‑
20min后弃上清;随后加入质量体积分数为2%的6
‑
氨基
‑3‑
吡啶苯硼酸室温搅拌1h后,磁吸5
‑
20min后弃上清,pb沉淀冲洗3遍,沉淀pb7.4复溶,于4℃保存。
14.进一步,所述步骤(2)通过载体偶联苯硼酸形成硼酸交联剂复合物,具体包括以下操作:通过edc方法将羧基苯硼酸或者是氨基苯硼酸固定于蛋白、聚合物、纳米粒子等载体上,形成苯硼酸交联剂。
15.进一步,所述糖蛋白用pb7.5(0.01mol/l)稀释至合适浓度,取200μl加入玻璃管中并加入一定量的磁性纳米探针,37℃反应5
‑
20min;磁吸5
‑
20min后弃上清,并超声洗涤2
‑
3遍,随后加入800l苯硼酸交联剂,超声反应5
‑
20min后,测定平均水化动力学粒径变化,通过信号的变化进一步确定糖蛋白的含量。
16.进一步,所述通过溶液平均水化粒径最大变化量确定特异性免疫识别元件标记磁性载体的标记量。
17.进一步,所述通过溶液平均水化粒径最大变化量确定磁性纳米探针的用量。
18.进一步,所述通过溶液平均水化粒径最大变化量确定苯硼酸交联剂的用量。
19.与现有技术相比,本技术的技术方案具备以下有益效果:
20.1、该基于苯硼酸交联剂的糖蛋白动态光散射免疫方法,利用硼酸与顺式二醇结合的独特性质,设计了识别单元苯硼酸交联剂用于糖蛋白顺式二醇的识别,硼酸与顺式二醇结合的独特性质使识别分子苯硼酸可作为抗体替代物用于仿生免疫学分析,因此,可在节约一个抗体的基础上实现对顺式二醇物质(糖蛋白)的多价结合和高灵敏检测,从而大大减少了对配对抗体的需求。
21.2、该基于苯硼酸交联剂的糖蛋白动态光散射免疫方法,使用目标蛋白特异性免疫识别元件标记磁性载体形成的免疫探针作为动态光散射信号增强探针,苯硼酸交联剂和免疫探针以目标糖蛋白为桥梁形成“多层夹心”结构前后溶液的平均水化动力学粒径变化作为动态光散射信号输出,利用水化动力学直径变化测定反应待检样品中糖蛋白的含量,操作步骤简单,短时间即可实现对复杂样本中糖蛋白的高灵敏检测,所制备的免疫磁珠可以
将目标蛋白从复杂的样品基质中分离富集出来,有效地消除了样品基质对后续检测的干扰。
附图说明
22.图1为本发明方法原理示意图;
23.图2为甲胎蛋白基于fe304磁微球的动态光散射均相免疫分析的标准曲线;
24.图3为氨基端脑钠肽前体基于fe304磁微球的动态光散射均相免疫分析的标准曲线;
25.图4为促绒毛膜性腺激素基于fe304磁微球的动态光散射均相免疫分析的标准曲线。
具体实施方式
26.下面将结合本发明的实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
27.请参阅图1
‑
4,一种基于苯硼酸交联剂的糖蛋白动态光散射免疫方法,苯硼酸交联剂和免疫识别元件标记的磁性载体作为动态光散射信号增强探针以目标糖蛋白为桥梁形成“多层夹心”结构前后溶液的平均水化动力学粒径变化作为动态光散射信号输出,利用水化动力学直径变化测定反应待检样品中糖蛋白的含量,包括以下步骤:
28.(1)磁性探针:采用edc一步法将糖蛋白特异性免疫识别元件标记羧基化磁性载体表面,获得特异性免疫识别元件标记的磁性探针;
29.(2)硼酸交联剂:通过载体共价偶联苯硼酸形成硼酸交联剂复合物,用于对糖蛋白的识别;
30.(3)糖蛋白的检测:在待测目标物溶液中加入特异性免疫识别元件标记的磁性探针和苯硼酸交联剂,37℃反应15
‑
30min后于马尔文纳米粒度仪上测定溶液的平均水化动力学直径,利用水化动力学直径变化测定反应待检样品中糖蛋白的含量。
31.该方法针对目标糖蛋白的特异性免疫识别元件单克隆抗体或者适配体等;磁性载体为fe304纳米微球、金磁颗粒、硅包磁、磁珠等;硼酸交联剂为蛋白、纳米粒子、聚合物作为载体连接苯硼酸形成的复合物等;糖蛋白用pb7.5(0.01mol/l)稀释至合适浓度,取200μl加入玻璃管中并加入一定量的磁性纳米探针,37℃反应5
‑
20min;磁吸5
‑
20min后弃上清,并超声洗涤2
‑
3遍,随后加入800l苯硼酸交联剂,超声反应5
‑
20min后,测定平均水化动力学粒径变化,通过信号的变化进一步确定糖蛋白的含量。
32.步骤(1)中采用edc一步法将糖蛋白特异性免疫识别元件标记于磁性载体表面形成磁性免疫探针,特异性免疫识别元件标记的羧基化磁性载体的具体制备方法为:取磁性载体于ph6.0
‑
8.opb(0.01mol/l)缓冲液中,加入特异性免疫识别元件后室温下搅拌反应,1
‑
乙基
‑
(3
‑
二甲基氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edc)重复加3次,反应完成磁吸5
‑
20min后弃上清;随后加入质量体积分数为2%的酸水解酪蛋白,并加入edc,室温搅拌1h后,磁吸5
‑
20min后弃上清;随后加入质量体积分数为2%的6
‑
氨基
‑3‑
吡啶苯硼酸室温搅拌1h
后,磁吸5
‑
20min后弃上清,pb沉淀冲洗3遍,沉淀pb7.4复溶,于4℃保存。
33.在该优选技术方案中:步骤(1)用于免疫识别元件标记的磁性探针,为了获得高的灵敏度,免疫识别元件采用不同的标记量,最终免疫识别元件的标记量通过与目标物及苯硼酸交联剂孵育得到最大的粒径增加量信号确定;免疫识别元件标记的磁性载体的最终用量通过设置不同的反应投入量,反应完成后测定溶液的平均水化动力学直径,得到最大的粒径增加量信号确定磁性纳米探针的最佳使用浓度。
34.步骤(2)通过载体偶联苯硼酸形成硼酸交联剂复合物,具体包括以下操作:通过edc方法将羧基苯硼酸或者是氨基苯硼酸固定于蛋白、聚合物、纳米粒子等载体上,形成苯硼酸交联剂。
35.步骤(3)中作为苯硼酸交联剂的用量,在免疫识别元件标记的磁性探针的标记量及磁性纳米探针确定后,通过与目标物及磁性纳米探针孵育得到最大的粒径增加量信号来确定。
36.各缓冲液及复溶液在使用前均需经0.22μm滤膜过滤后再使用。
37.需要说明的是,本方法适用于具有糖基化的蛋白,特别是适合于目标分析物的痕量检测。
38.该方法中信号输出底物为磁性载体,1)当溶液中糖蛋白含量为零或者极低时,磁性载体上的免疫识别元件不能或不与目标抗原结合,进而苯硼酸交联剂不能与糖蛋白结合,因此不能形成磁性纳米探针
‑
抗原
‑
苯硼酸交联剂三合一的“多层夹心”结构,此时溶液的平均水化动力学直径较空白值(标记了免疫识别元件的磁性载体)增加较小或不增加;2)随着溶液中糖蛋白含量增加时,磁性载体上的免疫识别元件与目标抗原结合,进而苯硼酸交联剂能与糖蛋白结合因此形成磁性纳米探针
‑
抗原
‑
苯硼酸交联剂三合一的“多层夹心”结构所占比例逐渐增加,此时溶液的平均水化动力学直径较空白值(标记了免疫识别元件的磁性载体)增加越来越多;3)随着糖蛋白含量的变化,溶液的平均水化动力学直径增加量出现线性变化,最终实现对糖蛋白的定量、灵敏检测。
39.磷酸盐缓冲液(pbs,0.05m,ph7.4)的配置方法:nacl40g,na2hp04 13.5g,kh2p04 1.0g,kcl 1.0g溶于1l超纯水中。
40.实施例1利用磁性纳米微球检测甲胎蛋白含量的应用
41.1羧基化fe304磁微球的制备
42.1)油酸化fe304磁珠的制备
43.a.fe304磁珠的合成,在500ml三口烧瓶中加入300ml超纯水,通入n2以除去水中氧气,同时在50℃预热20min,加入3.2gfecl2
·
h20,5.2gfecl3磁力搅拌混匀后加入25ml氨水,黄色溶液迅速变为黑色,50℃恒温反应30min,将合成的磁珠通过磁吸分离,用超纯水洗3
‑
5次直至磁吸后溶液ph至中性后复溶于300ml超纯水中。
44.b.fe304磁珠的油酸化修饰,将上步所合磁珠加入三口烧瓶中,通入n2,边搅拌边逐滴加入2.4ml油酸,由于油酸化的磁珠疏水,吸附于搅拌器或烧瓶壁上,70℃反应3h黑色溶液逐渐清澈,停止反应,倒掉烧杯中溶液,加入300ml乙醇将吸附的油酸化磁珠洗脱,磁吸3min弃去乙醇溶剂,重复此步骤4次直至乙醇液面无漂浮油酸层。
45.2)羧基化化fe304磁微球的合成
46.5mg聚马来酸酐
‑1‑
十八烯双亲链和10mg粒径10nm左右的油酸化fe304磁珠置于进
样瓶中,加入120l氯仿,涡旋充分混匀后加入250l的10mg/ml十二烷基硫酸钠水溶液,将上述混合物超声乳化,超声参数设置为:功率8%,时间2min,超声5s间隔10s,超声乳化结束后置于60℃烘箱中4h以挥发氯仿,13500rpm离心15min,最后将合成的苯硼酸化fe304磁微球载体悬浮于1ml超纯水中备用,估计离心损失率为20%,所以得到的苯硼酸化fe304磁微球载体浓度为12mg/ml。
47.2检测抗体标记的fe304微球探针的制备
48.取10μl上述合成的羧基化的fe304纳米微球加入到200μl ph8.0pb,加入3μg甲胎蛋白对应的检测抗体,室温下搅拌反应30min;继续搅拌反应,加入0.2μg 1
‑
乙基
‑
(3
‑
二甲基氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐后,室温搅拌30min,补加两次,磁吸5min后弃上清,随后加入质量体积分数为2%的酸水解酪蛋白,并加入0.2μg 1
‑
乙基
‑
(3
‑
二甲基氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐,室温搅拌1h后,磁吸5min后弃上清,随后加入质量体积分数为2%的6
‑
氨基
‑3‑
吡啶苯硼酸室温搅拌1h后,磁吸5min后弃上清,pb沉淀冲洗3遍,沉淀pb7.4复溶于4℃保存。
49.3苯硼酸交联剂的制备
50.通过8arm peg
‑
琥珀酰亚胺戊二酸酯和3
‑
氨基苯基硼盐酸盐(3
‑
apba)的共价偶联合成了8arm peg 3
‑
apba复合物作为苯硼酸交联剂,详细地,在4ml pb7.4(0.01m)溶液中添加166.08mg的3
‑
apba,并将溶液的ph调节至碱性,然后,将800mg 8arm peg
‑
琥珀酰亚胺戊二酸酯溶解于16ml pb7.4(0.01m)中,然后将其添加至上述溶解的3
‑
apba溶液中在室温下搅拌反应4h,反应完成后,将制备的8arm peg@3
‑
apba在0.01m pbs中透析3天,最后,对获得的8arm peg@3
‑
apba进行定量,并于4℃保存。
51.4检测甲胎蛋白的含量
52.本发明新型动态光散射免疫检测方法用于检测甲胎蛋白含量时,通过以下步骤实施:用本发明检测方法进行检测、分析结果。
53.1)将在医院通过化学发光定量好的甲胎蛋白抗原阳性样本分别用pbs7.4稀释,浓度按照所需的实际检测限确定;
54.2)稀释好的甲胎蛋白抗原放入4℃冰箱备用。
55.3)用本发明检测方法进行糖蛋白的含量。
56.4)分析结果。
57.用上述制备的15个不同浓度的标准品500、250、125、62.5、31.3、15.6、7.8、3.9、1.95、0.98、0.49、0.245、0.12、0.06和0pg/ml,在马尔文纳米粒度分析仪上测试溶液对应的平均水化动力学直径。
58.以溶液粒径为纵坐标,以甲胎蛋白浓度(pg/ml)为横坐标绘制标准曲线,求出线性方程,当进行实际样本检测时,将样本的粒径增加值代入标准曲线中,从标准曲线上读出所对应样本的浓度,乘以其对应的稀释倍数即为样本中甲胎蛋白的实际浓度。
59.实施例2糖蛋白
‑
甲胎蛋白作为待检物
60.6μl甲胎蛋白检测抗体标记的fe304微球探针与200μl不同浓度的甲胎蛋白样本反应5min,磁吸5min弃上清并用pb7.5(0.01mol/l)超声冲洗2
‑
3遍,随后加入800μl 0.2mg/ml的8arm peg@3
‑
apba复溶并超声,25℃孵育5min后,取出于25℃下用马尔文纳米粒度仪测定溶液的平均水化动力学直径变化,通过计算平均值后代入到标准曲线获得待检样品中甲胎
蛋白的浓度,具体实验结果如下:线性标准曲线为y=19.68ln(x) 246,r=0.9875,见附图2,该方法的最低检测限被定义为20个第一个标准(0标准时溶液的平均水化粒径)时平均水化粒径 3倍标准偏差(3倍的第一个标准样品三个平行样品的标准偏差),所需的抗原浓度,通过该标准曲线计算得最低检测线为0.33pg/ml。
61.实施例3利用金磁微球检测甲胎蛋白含量的应用
62.1检测抗体标记的金磁探针的制备
63.取10μl金磁纳米粒子(10mg/ml)加入到200μl ph8.0pb,加入4μg甲胎蛋白对应的检测抗体,室温下搅拌反应30min;继续搅拌反应,加入0.2μg 1
‑
乙基
‑
(3
‑
二甲基氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐后,室温搅拌30min,补加两次,反应完成后磁吸5min弃上清,随后加入质量体积分数为2%的酸水解酪蛋白,并加入0.2μg 1
‑
乙基
‑
(3
‑
二甲基氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐,室温搅拌1h后,磁吸5min后弃上清,随后加入质量体积分数为2%的6
‑
氨基
‑3‑
吡啶苯硼酸室温搅拌1h后,磁吸5min后弃上清,pb沉淀冲洗3遍,沉淀pb7.4复溶于4℃保存。
64.2检测甲胎蛋白的含量
65.本发明新型动态光散射免疫检测方法用于检测甲胎蛋白含量时,通过以下步骤实施:用本发明检测方法进行检测、分析结果。
66.1)将在医院通过化学发光定量好的甲胎蛋白抗原阳性样本分别pbs7.4稀释,浓度按照所需的实际检测限确定;
67.2)稀释好的甲胎蛋白抗原放入4℃冰箱备用。
68.3)用本发明检测方法进行糖蛋白的含量。
69.4)分析结果。
70.用上述制备的15个不同浓度的标准品500、250、125、62.5、31.3、15.6、7.8、3.9、1.95、0.98、0.49、0.245、0.12、0.06和0pg/ml,在马尔文纳米粒度分析仪上测试溶液对应的平均水化动力学直径。
71.以溶液粒径为纵坐标,以甲胎蛋白浓度(pg/ml)为横坐标绘制标准曲线,求出线性方程,当进行实际样本检测时,将样本的粒径增加值代入标准曲线中,从标准曲线上读出所对应样本的浓度,乘以其对应的稀释倍数即为样本中甲胎蛋白的实际浓度。
72.实施例4糖蛋白
‑
人绒毛膜促性腺激素作为待检物
73.6μl人绒毛膜促性腺激素检测抗体标记的fe304微球探针与200μl不同浓度的人绒毛膜促性腺激素反应5min,磁吸5min弃上清并用pb7.5(0.01mol/l)超声冲洗2
‑
3遍,随后加入800μl0.2mg/ml的8arm peg@3
‑
apba复溶并超声,25℃孵育5min后,取出于25℃下用马尔文纳米粒度仪测定溶液的平均水化动力学直径变化,通过计算平均值后代入到标准曲线获得待检样品中人绒毛膜促性腺激素的浓度,具体实验结果如下:线性标准曲线为y=11.774ln(x) 252.1,r=0.9845,见附图2,该方法的最低检测限被定义为20个第一个标准(0标准时溶液的平均水化粒径)时平均水化粒径 3倍标准偏差(3倍的第一个标准样品三个平行样品的标准偏差),所需的抗原浓度,通过该标准曲线计算得最低检测线为0.15mlu/ml。
74.实施例5糖蛋白
‑
氨基末端脑钠肽前体作为待检物
75.6μl氨基末端脑钠肽前体检测抗体标记的fe304微球探针与200μl不同浓度的氨基
末端脑钠肽前体反应5min,磁吸5min弃上清并用pb7.5(0.01mol/l)超声冲洗2
‑
3遍,随后加入800μl 0.2mg/ml的8arm peg@3
‑
apba复溶并超声,25℃孵育5min后,取出于25℃下用马尔文纳米粒度仪测定溶液的平均水化动力学直径变化,通过计算平均值后代入到标准曲线获得待检样品中氨基末端脑钠肽前体的浓度,具体实验结果如下:线性标准曲线为y=15.608ln(x) 284.91,r=0.9881,见附图2,该方法的最低检测限被定义为20个第一个标准(0标准时溶液的平均水化粒径)时平均水化粒径 3倍标准偏差(3倍的第一个标准样品三个平行样品的标准偏差),所需的抗原浓度,通过该标准曲线计算得最低检测线为14fg/ml。
76.实施例6bsa@cpba作为苯硼酸交联剂的合成
77.牛血清白蛋白(bsa)与羧基苯硼酸(cpba)的偶联是通过edc方法合成的,首先,将31.43mg的cpba溶解于1.0ml的n,n
‑
二甲基甲酰胺(dmf)中,加入108.92mg 1
‑
乙基
‑
(3
‑
二甲基氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edc)和130.8mg n
‑
羟基琥珀酰亚胺(nhs)在室温下搅拌6h,然后,将250mg bsa溶于10ml pb 7.5(0.01m)中,然后加入上述活化的cpba溶液中,在室温下搅拌过夜,将ph调节至7.5,最后,将制备的bsa@cpba在0.01m pbs中透析3天,并保存在4℃备用。
78.实施例7 cuncs@mba作为苯硼酸交联剂的合成
79.cuncs是根据以前报道的方法合成的,首先,将32mg巯基苯硼酸(mba)溶解在包含7.0ml dmf和1.0ml去离子水的混合溶液中,超声处理40min,将溶液用0.22μm过滤器过滤,然后,将5.0mg cus04溶解在0.5ml去超纯水,并添加到溶解的mba溶液中在室温下搅拌3小时,并将合成的cuncs@pba保存在4℃备用。
80.实施例8 si02@3
‑
apba作为苯硼酸交联剂的合成
81.si02@3
‑
apba复合物是通过si02和3
‑
氨基苯硼酸盐酸盐(3
‑
apba)的共价偶联而合成的,将200μl 10mg/ml sio2溶解在2.0ml pb 6.0(0.01m)中,随后加入150l 50mg/ml 3
‑
apba并将ph值调节至6.0,在室温下搅拌30min,随后,将25g edc添加到上述溶液中,并重复3次,反应完成后,15000rpm离心15min,然后用pb 7.4(0.01m)洗涤两次,最后将沉淀物重悬于500μl pb ph7.4(0.01m)中,并保存于4℃备用。
82.需要说明的是,在本文中,诸如第一和第二等之类的关系术语仅仅用来将一个实体或者操作与另一个实体或操作区分开来,而不一定要求或者暗示这些实体或操作之间存在任何这种实际的关系或者顺序。而且,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。在没有更多限制的情况下,由语句“包括一个
……”
限定的要素,并不排除在包括所述要素的过程、方法、物品或者设备中还存在另外的相同要素。
83.尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。