一种基于腔肠素
‑
h的氘代化合物及其制备方法与应用
技术领域
1.本发明属于药物技术领域,涉及萤光素酶底物的制备,具体涉及一种基于腔肠素
‑
h(coelenterazine
‑
h)的氘代化合物及其制备方法与应用。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.生物发光(bioluminescene)是一种在活体生物上进行发光的特殊现象,由萤光素酶与其底物萤光素的相互作用引起的,萤光素酶通过分子氧催化萤光素氧化,以激发态转化为氧化萤光素分子,氧化萤光素发出可见光而返回基态。所有已知生物发光体系产生的光谱范围在400至700nm之间。许多底栖、浅水和深水生物产生蓝色发光,但是在特殊情况下某些发光生物会产生不同的颜色。
4.在海洋系统中使用最为广泛的萤光素是腔肠素(coelenterazine)。大多数发光的海洋生物都可与腔肠素产生生物发光。腔肠素是一种修饰的双环三肽,由两个酪氨酸残基和一个苯丙氨酸残基构成。最初因其存在于刺胞aequorea和renilla中而得名,后来发现在许多生物中都存在此分子。腔肠素
‑
h是腔肠素改造过程中,得到的比较优秀的类似物。萤光素酶与腔肠素共同存在于自然界,目前已从腔肠素生物发光系统中分离出许多天然萤光素酶并进行测序,但在这些不同的萤光素酶中,仅renilla、gaussia和metridia longa萤光素酶得到广泛的应用。海肾(renilla)萤光素酶是最早被克隆的萤光素酶,其质量为36kda,由311个氨基酸组成,生物发光最大发射波长λ
max
为480nm。这种萤光素酶几乎可以在所有细胞类型中表达,目前在生物成像和其他生物发光研究中最受欢迎。除天然rluc外,还产生了具有增强的稳定性、亮度甚至红移生物发光光谱的突变型萤光素酶。
5.在生物医学中,生物发光已经应用于各种疾病的研究,例如癌症、心脏病、神经退行性疾病和传染病等,为人类的健康事业做出重要贡献。生物发光还用于创建响应于某些分析物而发光的经修饰的细胞系统,因此已经产生非常灵敏的生物传感器;生物发光也作为非常灵敏的技术应用于免疫测定、atp测定和实时生物发光测定。生物发光在医学界的主要应用是生物发光成像技术和生物发光共振能量转移。但是生物发光体系存在一些问题,例如生物发光强度低,持续时间短,生物发光的发射波长较短,对组织伤害较大等问题。因此如何改善这些缺点是学者亟需解决的问题,而萤光素作为小分子底物,增加改造的简单性和可行性,因此对萤光素酶底物的改造可以推动生物发光在成像领域的应用,更甚推动在生物发光在生命领域的应用。
技术实现要素:
6.为了解决现有技术的不足,本发明的目的是提供一种基于腔肠素
‑
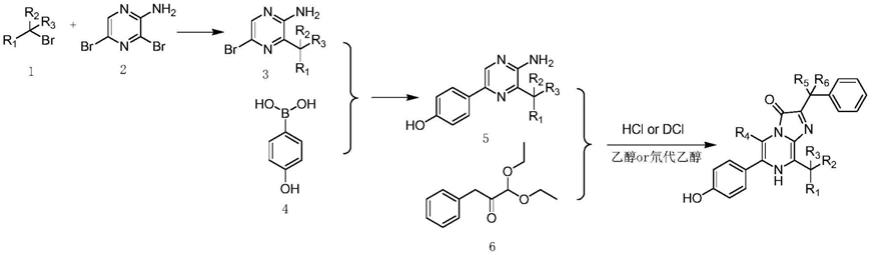
h的氘代化合物及其制备方法与应用,通过对腔肠素
‑
h的c
‑
2、c
‑
5、c
‑
8位进行氘代设计,比腔肠素
‑
h具有更
好的生物发光强度。本发明的氘代化合物可以作为腔肠素类生物发光系统的底物,合成方法简单,合成路线经典、经济。
7.为了实现上述技术目的,本发明的技术方案为:
8.第一方面,一种基于腔肠素
‑
h的氘代化合物,化学结构式如下:
[0009][0010]
其中,r1为苯基或氘代苯基,r2为氢或氘,r3为氢或氘,r4为氢或氘,r5为氢或氘,r6为氢或氘,r1~r6中的至少一个基团为氘代基团。
[0011]
本发明对腔肠素
‑
h的c
‑
2、c
‑
5、c
‑
8位进行氘代,一方面,形成氘代化合物会保留腔肠素
‑
h原来的生物活性和选择性;另一方面,氘原子的重量是氢原子的两倍,携带中子的氘与碳形成的碳一氘键以较低的频率振动。因而,本发明对腔肠素
‑
h的c
‑
2、c
‑
5、c
‑
8位进行氘代后,碳氘键比碳氢键稳定,用氘原子取代化合物中的氢原子可直接影响某些化合物的吸收、分布、代谢和排泄等属性,缓解其分解的过程,延长氘代化合物在体内的作用时间,达到改变化合物代谢速度与代谢途径的目的,从而增加生物发光强度和减缓生物发光衰减速度。
[0012]
本发明所述氘代苯基可以为一氘代苯基、二氘代苯基、三氘代苯基、四氘代苯基、五氘代苯基。其中,一氘代苯基可以为2
‑
氘代苯基、3
‑
氘代苯基、4
‑
氘代苯基,二氘代苯基可以为2,3
‑
二氘代苯基、2,4
‑
二氘代苯基、2,5
‑
二氘代苯基、2,6
‑
二氘代苯基、3,4
‑
二氘代苯基、3,5
‑
二氘代苯基,三氘代苯基可以为2,3,4
‑
三氘代苯基、2,3,5
‑
三氘代苯基、2,3,6
‑
三氘代苯基、2,4,5
‑
三氘代苯基、2,4,6
‑
三氘代苯基、2,5,6
‑
三氘代苯基、3,4,5
‑
三氘代苯基,四氘代苯基可以为2,3,4,5
‑
四氘代苯基、2,3,4,6
‑
四氘代苯基、2,3,5,6
‑
四氘代苯基。在一些实施例中,氘代苯基为五氘代苯基。
[0013]
r2~r6可以选择部分相同,也可以选择全部相同。在一些实施例中,r2~r6全部相同。
[0014]
在一些实施例中,包括以下化合物:
[0015]2‑
(1,1
‑
二氘
‑1‑
苯基甲基)
‑5‑
氘
‑6‑
(4
‑
羟基苯基)
‑8‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)咪唑[1,2
‑
a]吡嗪
‑
3(7h)
‑
酮(c1);
[0016]2‑
苄基
‑6‑
(4
‑
羟基苯基)
‑8‑
((2,3,4,5,6
‑
五氘苯基)甲基)咪唑[1,2
‑
a]吡嗪
‑
3(7h)
‑
酮(c2);
[0017]5‑
氘
‑6‑
(4
‑
羟基苯基)
‑
2,8
‑
二(1,1
‑
二氘
‑1‑
苯基甲基)咪唑[1,2
‑
a]吡嗪
‑
3(7h)
‑
酮(c3)。
[0018]
另一方面,一种基于腔肠素
‑
h的氘代化合物的制备方法,包括如下反应路线制备获得;
[0019][0020]
其中,r1为苯基或氘代苯基,r2为氢或氘,r3为氢或氘,r4为氢或氘,r5为氢或氘,r6为氢或氘,r1~r6中的至少一个基团为氘代基团。
[0021]
在一些实施例中,化合物1与化合物2进行偶联反应获得化合物3。所述偶联反应优选negishi反应。在一种或多种实施例中,获得化合物3的过程为,先将化合物1与锌粉、碘单质反应形成有机锌试剂,再将有机锌试剂与化合物2进行negishi反应。具体地,形成有机锌试剂的温度为82~88℃,反应时间为3~5h。具体地,negishi反应的温度为室温,反应时间为12~24h。本发明所述的室温是指室内环境的温度,一般为15~30℃。negishi反应的催化剂优选为双三基磷二氯化钯。化合物1与化合物2的摩尔比为1:0.6~0.8。化合物1与锌粉、碘单质的摩尔比为1:1.5~3.5:0.06~0.09。
[0022]
在一些实施例中,化合物3与化合物4通过suzuki偶联反应获得化合物5。在一种或多种实施例中,反应温度为75~85℃,反应时间为1~2h。化合物3与化合物4的摩尔比为1:1.4~1.6。suzuki偶联反应的催化剂优选为双三苯基膦二氯化钯。
[0023]
在一些实施例中,化合物5与化合物6进行环化反应获得基于腔肠素
‑
h的氘代化合物。在一种或多种实施例中,反应温度为80~90℃,反应时间为8~12h。化合物5与化合物6的摩尔比为1:1.8~2.2。
[0024]
在一些实施例中,化合物6由二乙氧基乙酸乙酯与苄基氯化镁反应获得。其反应式如下所示:
[0025][0026]
在一种或多种实施例中,反应温度为
‑
79~
‑
77℃,反应时间为2~5h。二乙氧基乙酸乙酯与苄基氯化镁摩尔比为1:1.4~1.6。
[0027]
更为具体地,提供c1、c2和c3的制备方法,反应式如下:
[0028][0029][0030]
试剂和条件:(i)nbs,过氧化苯甲酸叔丁酯,四氯化碳,90℃,2h;(ii)碘,活化锌粉,四氢呋喃,n,n
‑
二甲基甲酰胺,85℃,3h;1,2
‑
二溴乙烷,室温,24h;(iii)双三苯基膦二氯化钯,碳酸钾,甲苯,水,乙醇,80℃,1.5h;(iv)四氢呋喃,
‑
78℃,2h;(v)35%氯化氘,氘代乙醇,8h;(vi)浓盐酸,乙醇,8h。
[0031]
步骤如下:
[0032]
(1)以氘代甲苯为原料与n
‑
溴代琥珀酰亚胺进行反应,得中间体苄溴
‑
d7;
[0033]
(2)苄溴
‑
d7与2
‑
氨基
‑
3,5
‑
二溴吡嗪反应,得中间体2
‑
氨基
‑3‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)
‑5‑
溴
‑
吡嗪;
[0034]
(2’)苄溴与2
‑
氨基
‑
3,5
‑
二溴吡嗪反应,得中间体2
‑
氨基
‑3‑
苄基
‑5‑
溴吡嗪;
[0035]
(3)2
‑
氨基
‑3‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)
‑5‑
溴
‑
吡嗪与4
‑
羟基苯硼酸反应,得中间体2
‑
氨基
‑3‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)
‑5‑
(4
‑
羟基苯基)
‑
吡嗪;
[0036]
(3’)2
‑
氨基
‑3‑
苄基
‑5‑
溴吡嗪与4
‑
羟基苯硼酸反应,得中间体2
‑
氨基
‑3‑
苄基
‑5‑
(4
‑
羟基苯基)
‑
吡嗪;
[0037]
(4)以二乙氧基乙酸乙酯为原料,与苄基氯化镁反应,得中间体1,1
‑
二乙氧基
‑3‑
苯基丙
‑2‑
酮;
[0038]
(5)以2
‑
氨基
‑3‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)
‑5‑
(4
‑
羟基苯基)
‑
吡嗪和1,1
‑
二乙氧基
‑3‑
苯基丙
‑2‑
酮为原料,在35%氯化氘的作用下,得化合物c1;
[0039]
(5’)以2
‑
氨基
‑3‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)
‑5‑
(4
‑
羟基苯基)
‑
吡嗪和1,1
‑
二乙氧基
‑3‑
苯基丙
‑2‑
酮为原料,在浓盐酸的作用下,得化合物c2;
[0040]
(5”)以2
‑
氨基
‑3‑
苄基
‑5‑
(4
‑
羟基苯基)
‑
吡嗪和1,1
‑
二乙氧基
‑3‑
苯基丙
‑2‑
酮为原料,在35%氯化氘的作用下,得化合物c3。
[0041]
步骤(1)中,溶剂为四氯化碳;反应温度为80~110℃,反应时间为2~4h;所述氘代甲苯与n
‑
溴代琥珀酰亚胺摩尔比为1:(0.8~1.2)。
[0042]
步骤(2)中,溶剂为超干四氢呋喃(h2o≤50ppm):超干n,n
‑
二甲基甲酰胺=5:1(v/v);溶解2
‑
氨基
‑
3,5
‑
二溴吡嗪的溶剂超干n,n
‑
二甲基甲酰胺,合成锌试剂的温度为85℃;反应时间为3~5h,锌试剂与2
‑
氨基
‑
3,5
‑
二溴吡嗪反应的温度为室温,反应时间为12~24h;苄溴
‑
d7、活化锌粉、碘单质、1,2
‑
二溴乙烷、2
‑
氨基
‑
3,5
‑
二溴吡嗪与双三基磷二氯化钯摩尔比为1:1.5:0.08:0.09:0.7:0.03。
[0043]
步骤(2’)中,溶剂为超干四氢呋喃:超干n,n
‑
二甲基甲酰胺=5:1(v/v);溶解2
‑
氨基
‑
3,5
‑
二溴吡嗪的溶剂超干n,n
‑
二甲基甲酰胺,合成锌试剂的温度为85℃;反应时间为3~5h,锌试剂与2
‑
氨基
‑
3,5
‑
二溴吡嗪反应的温度为室温,反应时间为12~24h;苄溴、活化锌粉、碘单质、2
‑
氨基
‑
3,5
‑
二溴吡嗪与双三基磷二氯化钯摩尔比为1.3:4:0.1:1:0.1。
[0044]
步骤(3)中,溶剂为甲苯、乙醇、水的混合物;甲醇、乙醇、水的体积比为2:4:1;反应温度为80℃,反应时间为1~2h;2
‑
氨基
‑3‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)
‑5‑
溴
‑
吡嗪、4
‑
羟基苯硼酸、双三苯基膦二氯化钯与碳酸钾摩尔比为1:1.5:(0.03~0.05):(2~3)
[0045]
步骤(3’)中,溶剂为甲苯、乙醇、水的混合物;甲醇、乙醇、水的体积比为2:4:1;反应温度为80℃,反应时间为1~2h;2
‑
氨基
‑3‑
苄基
‑5‑
溴
‑
吡嗪、4
‑
羟基苯硼酸、双三苯基膦二氯化钯与碳酸钾摩尔比为1:1.5:(0.03~0.05):(2~3)。
[0046]
步骤(4)中,溶剂为四氢呋喃,优选为超干四氢呋喃;反应温度零下78℃;反应时间为2~5h;二乙氧基乙酸乙酯与苄基氯化镁摩尔比为1:1.5。
[0047]
步骤(5)中,溶剂为氘代乙醇(etod);反应温度为85℃,反应时间8~12h;2
‑
氨基
‑3‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)
‑5‑
(4
‑
羟基苯基)
‑
吡嗪、1,1
‑
二乙氧基
‑3‑
苯基丙
‑2‑
酮、35%氯化氘摩尔比为1:2:10。
[0048]
步骤(5’)中,反应溶剂为乙醇;反应温度为85℃,反应时间8~12h;2
‑
氨基
‑3‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)
‑5‑
(4
‑
羟基苯基)
‑
吡嗪、1,1
‑
二乙氧基
‑3‑
苯基丙
‑2‑
酮、浓盐酸摩尔比为1:2:5。本发明所述浓盐酸为质量份数为35~36%的盐酸。
[0049]
步骤(5”)中,反应溶剂为氘代乙醇(etod);反应温度为85℃,反应时间8~12h;2
‑
氨基
‑3‑
苄基
‑5‑
(4
‑
羟基苯基)
‑
吡嗪、1,1
‑
二乙氧基
‑3‑
苯基丙
‑2‑
酮、35%氯化氘摩尔比为1:2:10。
[0050]
第三方面,上述基于腔肠素
‑
h的氘代化合物的以下任一应用;
[0051]
(1)作为生物发光底物中的应用;
[0052]
(2)作为探针在检测萤光素酶的应用;
[0053]
(3)在萤光酶的存在下作为报告信号检测药物在酶水平、细胞水平和/或动物水平的药理和毒性的应用。
[0054]
本发明所述优选的,以非疾病的诊断与治疗为目的。
[0055]
第四方面,一种生物发光探针,包括上述基于腔肠素
‑
h的氘代化合物。
[0056]
第五方面,一种荧光酶检测试剂盒,包括上述基于腔肠素
‑
h的氘代化合物、缓冲溶液。
[0057]
本发明的有益效果为:
[0058]
经实验证明,本发明中的基于腔肠素
‑
h的氘代化合物与酶水平、细胞水平,动物水平有良好的生物活性作用;在酶水平表现出化合物浓度依赖性,化合物c1和c3的生物发光强度优于腔肠素
‑
h,在米氏常数k
m
方面,c2表现突出;在细胞水平,表现出化合物浓度和细胞浓度依赖性,化合物c1、c2、c3的生物发光强度优于腔肠素
‑
h;在动物水平,c1、c2、c3的生物发光强度优于腔肠素
‑
h。
附图说明
[0059]
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
[0060]
图1为本发明实施例4的海肾萤光素酶的体外酶活性检测的结果图,a为腔肠素
‑
h的氘代物与rluc作用后的成像结果,b为腔肠素
‑
h的氘代物与rluc作用后,生物发光随化合物浓度的变化;
[0061]
图2为本发明实施例5的细胞es
‑
2的活性的结果图,(a)腔肠素
‑
h的氘代物与稳定表达rluc的es
‑
2细胞作用后的成像结果图;(b)腔肠素
‑
h的氘代物与稳定表达rluc的es
‑
2细胞作用后,生物发光随化合物浓度的变化;(c)腔肠素
‑
h的氘代物(5μm)与稳定表达rluc的es
‑
2细胞作用后,生物发光随时间的变化;
[0062]
图3为本发明实施例6的动物水平的生物发光结果图,(a)在1mm时,腔肠素
‑
h的氘代物在裸鼠上生物发光成像结果图;(b)1mm时,147
‑
57、147
‑
60和147
‑
61与皮下荷瘤裸鼠成像结果随时间的变化;(c)1mm时,147
‑
57、147
‑
60和147
‑
61腔肠素
‑
h与皮下荷瘤裸鼠的成像结果量化图。
具体实施方式
[0063]
应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
[0064]
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
[0065]
为了使得本领域技术人员能够更加清楚地了解本发明的技术方案,以下将结合具
体的实施例详细说明本发明的技术方案。
[0066]
实施例1:2
‑
(1,1
‑
二氘
‑1‑
苯基甲基)
‑5‑
氘
‑6‑
(4
‑
羟基苯基)
‑8‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)咪唑[1,2
‑
a]吡嗪
‑
3(7h)
‑
酮(c1)的制备。
[0067]
1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)
‑1‑
溴甲烷的制备:
[0068]
氘代甲苯(1g,9.98mmol,toluene
‑
d8)和n
‑
溴代琥珀酰亚胺(1.78g,9.98mmol)溶于50ml的四氯化碳中,然后加入过氧化苯甲酸叔丁酯(581mg,2.99mmol)。在90℃的条件下反应2h,使用tlc监测反应,反应完全,停止反应,过滤,除去反应液中的丁二酰亚胺,收集滤液,旋干,加入100
‑
200目硅胶拌样,以200
‑
300目硅胶装柱,以石油醚为洗脱剂,得中间体化合物1,r
f
约为0.5,展开剂:石油醚,无色油状化合物1.1g,产率为62%。
13
c nmr(101mhz,dmso)δ138.22,129.56,129.32,129.08,128.88,128.64,128.53,128.39,128.29,128.05,40.67,40.46,40.25,40.04,39.83,39.62,39.42,34.97,34.73,34.50,34.27,34.04.
[0069]2‑
氨基
‑3‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)
‑5‑
溴
‑
吡嗪的制备:
[0070]
活化的锌粉(1.82g,27.85mmol)加入到50ml的双颈瓶中,在氮气保护下、室温条件下,加入碘单质(212mg,0.84mmol)溶液(碘单质溶于超干四氢呋喃和超干n,n
‑
二甲基甲酰胺,超干四氢呋喃:n,n
‑
二甲基甲酰胺=5:1),搅拌至碘单质的颜色褪去。使用注射器将1,1
‑
二氘
‑1‑
溴
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲烷(2.48g,13.93mmol)和1,2
‑
二溴乙烷(69mg,0.37mmol)加入到上述的溶液中,85℃条件下回流反应。经3h反应后,反应液(呈灰色的均一的溶液)静止冷却至室温。将2
‑
氨基
‑
3,5
‑
二溴吡嗪(2.35g,9.28mmol)和双三苯基膦二氯化钯(195mg,0.28mmol)溶于超干n,n
‑
二甲基甲酰胺溶液中,将其滴加到上述溶液中。室温条件下,反应24h。反应终止,将反应液经硅藻土过滤,在滤液中加入200ml的饱和氯化铵溶液和2
×
200ml的乙酸乙酯溶液萃取,收集有机层溶液加入无水硫酸钠干燥,过滤,收集滤液,旋干,加入100
‑
200目硅胶拌样,以200
‑
300目硅胶装柱,以乙酸乙酯和石油醚为洗脱剂,进行梯度洗脱,得中间体,r
f
=0.3,展开剂为石油醚:乙酸乙酯=3:1,淡黄色化合物1.3g,产率为52%。1h nmr(400mhz,dmso
‑
d6)δ7.95(s,1h),6.55(s,2h).esi
‑
ms:m/z[m h]
calcd for 271.06,272.05,found for 271.07,273.07.
[0071]2‑
氨基
‑3‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)
‑5‑
(4
‑
羟基苯基)
‑
吡嗪的制备:
[0072]2‑
氨基
‑3‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)
‑5‑
溴
‑
吡嗪(1g,4.80mmol)、4
‑
羟基苯硼酸(993mg,7.20mmol)、双三苯基膦二氯化钯(168mg,0.24mmol)和无水碳酸钾(1.33g,9.60mmol)溶于甲苯:乙醇:水=2:4:1的溶液里面,在氮气保护下,80℃的条件下,监测反应,至原料反应完全,停止反应,使用硅藻土过滤,在滤液中加入50ml饱和氯化铵溶液和2
×
50ml的乙酸乙酯,萃取,收集有机层溶液,无水硫酸钠干燥,过滤,收集滤液,旋干,加入100
‑
200目硅胶拌样,以200
‑
300目硅胶装柱,以乙酸乙酯和石油醚为洗脱剂,进行梯度洗脱,制备中间体,r
f
=0.3,展开剂为石油醚:乙酸乙酯=1:1,147
‑
40为淡黄色固体,产率为90%。熔点为214
‑
216℃。1h nmr(400mhz,dmso
‑
d6)δ9.50(s,1h),8.29(s,1h),7.73(d,j=8.4hz,2h),6.80(d,j=8.4hz,2h),6.21(s,2h).esi
‑
ms:m/z[m h]
calcd for 285.17,found for 285.32.
[0073]
1,1
‑
二乙氧基
‑3‑
苯基丙
‑2‑
酮的制备:
[0074]
2,2
‑
二乙氧基乙酸乙酯(2g,11.35mmol)溶于超干四氢呋喃,在氮气保护下,在零
下78℃条件下,保温,使用恒压滴定漏斗,滴加苄基氯化镁(2.57g,17.02mmol),30min内滴定完毕,零下78℃条件下,反应2小时,tlc监测反应,反应完毕,停止反应,转移至室温,迅速加入饱和氯化铵溶液(50ml)搅拌10分钟,加入乙酸乙酯(2
×
100ml),萃取,收集乙酸乙酯层溶液,无水硫酸钠干燥,过滤,收集滤液,旋干,加入100
‑
200目硅胶拌样,以200
‑
300目硅胶装柱,以乙酸乙酯:石油醚=20:1进行洗脱,得中间体147
‑
42,r
f
=0.8,展开剂为石油醚:乙酸乙酯=20:1,无色油状化合物1.6g,产率63%。1h nmr(400mhz,cdcl3)δ7.31(t,j=7.2hz,2h),7.22(t,j=7.9hz,3h),4.62(s,1h),3.89(s,2h),3.70(dq,j=9.4,7.1hz,2h),3.55(dq,j=9.5,7.0hz,2h),1.24(t,j=7.1hz,6h).esi
‑
ms:m/z[m h]
calcd for 237.15,found for 237.18.
[0075]2‑
(1,1
‑
二氘
‑1‑
苯基甲基)
‑5‑
氘
‑6‑
(4
‑
羟基苯基)
‑8‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)咪唑[1,2
‑
a]吡嗪
‑
3(7h)
‑
酮的制备:
[0076]2‑
氨基
‑3‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)
‑5‑
(4
‑
羟基苯基)
‑
吡嗪(100mg,0.35mmol)和1,1
‑
二乙氧基
‑3‑
苯基丙
‑2‑
酮(156mg,0.73mmol)溶于10ml的氘代乙醇(ethanol
‑
d)中,在氮气保护下,室温搅拌2h后,加入35%的氯化氘(376mg,3.5mmol),将反应液转移至80℃条件下,反应8h,冷却至室温,旋干,进行薄层层析,以二氯甲烷:甲醇=15:1为层析液,得目标色带,以二氯甲烷:甲醇=10:1为洗脱剂冲洗目标色带,得化合物147
‑
57,即为c1化合物,r
f
=0.3,展开剂为二氯甲烷:甲醇=15:1,淡黄色固体45mg,产率31%,熔点为145
‑
149℃。1h nmr(400mhz,meod)δ7.77(d,j=8.7hz,2h),7.48
–
7.17(m,5h),6.91(d,j=8.7hz,2h).
13
c nmr(101mhz,meod)δ
[0077]
159.07,144.57,143.03,137.50,135.73,128.21,128.02,126.54,126.32,123.66,115.49.esi
‑
hrms:m/z[m h]
calcd for 418.2334,found for 418.2332.
[0078]
化合物c1的化学结构式如下所示:
[0079][0080]
实施例2:2
‑
苄基
‑6‑
(4
‑
羟基苯基)
‑8‑
((2,3,4,5,6
‑
五氘苯基)甲基)咪唑[1,2
‑
a]吡嗪
‑
3(7h)
‑
酮(c2)的制备。
[0081]2‑
苄基
‑6‑
(4
‑
羟基苯基)
‑8‑
((2,3,4,5,6
‑
五氘苯基)甲基)咪唑[1,2
‑
a]吡嗪
‑
3(7h)
‑
酮的制备:
[0082]
实施例1制备的2
‑
氨基
‑3‑
(1,1
‑
二氘
‑1‑
(2,3,4,5,6
‑
五氘苯基)甲基)
‑5‑
(4
‑
羟基苯基)
‑
吡嗪(100mg,0.35mmol)和实施例1制备的1,1
‑
二乙氧基
‑3‑
苯基丙
‑2‑
酮(156mg,0.73mmol)溶于10ml的无水乙醇中,在氮气保护下,室温搅拌2小时后,加入浓盐酸(366mg,3.5mmol),将反应液转移至80℃条件下,反应8小时,冷却至室温,旋干,进行薄层层析,以二
氯甲烷:甲醇=15:1为层析液,得目标色带,以二氯甲烷:甲醇=10:1为洗脱剂冲洗目标色带,得化合物147
‑
60,即为c2化合物,r
f
=0.3,展开剂为二氯甲烷:甲醇=15:1,淡黄色固体45mg,产率为30%。熔点为154
‑
156℃。1h nmr(400mhz,meod)δ8.47(s,1h),7.78(d,j=8.7hz,2h),7.41
–
7.17(m,5h),6.92(d,j=8.7hz,2h),4.58(s,2h),4.31(s,2h).
13
c nmr(101mhz,meod)δ158.94,148.09,140.72,138.02,136.08,133.01,128.28,128.09,127.97,127.85,127.62,126.44,126.10,123.39,115.46,107.14,34.38,32.01.esi
‑
hrms:m/z[m h]
calcd for 413.2020,found for413.2056.
[0083][0084]
化合物c2的化学结构式如下所示:
[0085][0086]
实施例3:5
‑
氘
‑6‑
(4
‑
羟基苯基)
‑
2,8
‑
二(1,1
‑
二氘
‑1‑
苯基甲基)咪唑[1,2
‑
a]吡嗪
‑
3(7h)
‑
酮(c3)的制备。
[0087]2‑
氨基
‑3‑
苄基
‑5‑
溴
‑
吡嗪的制备:
[0088]
活化的锌粉(3.06g,46.77mmol)加入至50ml双颈瓶,在氮气保护、室温条件下,加入碘单质(356mg,1.4mmol)溶液(碘单质溶于超干四氢呋喃和超干n,n
‑
二甲基甲酰胺,超干四氢呋喃:n,n
‑
二甲基甲酰胺=5:1),搅拌,碘单质的颜色褪去。使用注射器将苄溴(4g,23.39mmol)加入到上述的溶液中,85℃回流反应。3h后,反应液(呈灰色的均一的溶液)静止冷却至室温。将2
‑
氨基
‑
3,5
‑
二溴吡嗪(3.94g,15.59mmol)和双三苯基膦二氯化钯(328mg,467.67μmol)溶于超干n,n
‑
二甲基甲酰胺溶液中,将其滴加到上述溶液中。室温条件下,反应24h。反应终止,反应液经硅藻土过滤,在滤液中加入200ml的饱和氯化铵溶液和2
×
200ml的乙酸乙酯溶液萃取,收集有机层溶液加入无水硫酸钠干燥,过滤,收集滤液,旋干,加入100
‑
200目硅胶拌样,以200
‑
300目硅胶装柱,以乙酸乙酯和石油醚为洗脱剂,进行梯度洗脱,得中间体,r
f
=0.3,展开剂为石油醚:乙酸乙酯=3:1,黄色油状化合物2.92g,产率71%。1h nmr(400mhz,dmso
‑
d6)δ7.94(s,1h),7.36
–
7.23(m,4h),7.20(t,j=6.9hz,1h),6.51(s,2h),3.97(s,2h).esi
‑
ms:m/z[m h]
calcd for 264.01,266.01,found for264.08,266.08.
[0089]2‑
氨基
‑3‑
苄基
‑5‑
(4
‑
羟基苯基)
‑
吡嗪的制备:
[0090]2‑
氨基
‑3‑
苄基
‑5‑
溴
‑
吡嗪(2g,7.57mmol)、4
‑
羟基苯硼酸(1.57g,11.36mmol)、双三苯基膦二氯化钯(200mg,0.28mmol)、无水碳酸钾(2.09g,15.14mmol)溶于甲苯:乙醇:水=2:4:1的溶液里面,在氮气保护下,80℃的条件下,监测反应,至原料反应完全,停止反应,使用硅藻土过滤,在滤液中加入50ml饱和氯化铵溶液和2
×
50ml的乙酸乙酯,萃取,收集有
机层溶液,无水硫酸钠干燥,过滤,收集滤液,旋干,加入100
‑
200目硅胶拌样,以200
‑
300目硅胶装柱,以乙酸乙酯和石油醚为洗脱剂,进行梯度洗脱,得中间体147
‑
20,r
f
=0.3,展开剂为石油醚:乙酸乙酯=1:1,淡黄色针状化合物2g,即为c2化合物,产率95%,熔点为220
‑
222℃。1h nmr(400mhz,dmso
‑
d6)δ9.48(s,1h),8.29(s,1h),7.73(d,j=8.3hz,2h),7.33(d,j=7.5hz,2h),7.27(dd,j=16.7,9.4hz,2h),7.19(t,j=7.0hz,1h),6.80(d,j=8.4hz,2h),6.19(s,2h),4.06(s,2h).esi
‑
ms:m/z[m h]
calcd for 278.13,found for 278.11.
[0091]5‑
氘
‑6‑
(4
‑
羟基苯基)
‑
2,8
‑
二(1,1
‑
二氘
‑1‑
苯基甲基)咪唑[1,2
‑
a]吡嗪
‑
3(7h)
‑
酮的制备:
[0092]2‑
氨基
‑3‑
苄基
‑5‑
(4
‑
羟基苯基)
‑
吡嗪(100mg,0.36mmol)和1,1
‑
二乙氧基
‑3‑
苯基丙
‑2‑
酮(200mg,0.90mmol)溶于10ml的氘代乙醇(ethanol
‑
d)中,在氮气保护下,室温搅拌2h后,加入35%的氯化氘(376mg,3.6mmol),将反应液转移至80℃条件下,反应8h,冷却至室温,旋干,进行薄层层析,以二氯甲烷:甲醇=15:1为层析液,得目标色带,以二氯甲烷:甲醇=10:1为洗脱剂冲洗目标色带,得化合物147
‑
61,即为c3化合物,r
f
=0.3,展开剂为二氯甲烷:甲醇=15:1,淡黄色固体50mg,熔点为145
‑
147℃,产率为34%。1h nmr(400mhz,meod)δ8.40(s,1h),7.71(d,j=8.1hz,2h),7.55
–
7.16(m,10h),6.86(d,j=8.1hz,2h),4.56(s,2h),4.31(s,2h).
13
c nmr(101mhz,meod)δ159.28,146.90,139.51,138.46,136.84,135.56,128.93,128.48,128.40,128.23,128.14,126.95,126.75,126.69,125.04,124.31,115.50,108.94,37.09,29.77.esi
‑
hrms:m/z[m h]
calcd for 408.1707.
[0093]
化合物c2的化学结构式如下所示:
[0094][0095]
实施例4:基于腔肠素
‑
h的氘代化合物与海肾萤光素酶(rluc)的体外酶活性研究
[0096]
氘代腔肠素
‑
h类化合物的浓储使用tris
‑
hcl缓冲液(50mm)分别稀释成50、20、10、4、2、1、0.5、0μm的化合物梯度,将海肾萤光素酶使用tris
‑
hcl缓冲液(50mm)稀释成1μg/ml,将50μl化合物分别加入全黑的96孔中,然后加入50μl海肾萤光素酶,每孔共100μl,混合均匀,迅速放入小动物活体成像仪,立即成像,曝光时间1s,binning为4,field of view为12.5,f
‑
stop为1,每30s成像一次,连续成像20min。利用graph pad软件的米氏方程模块,计算米氏常数(k
m
),酶促反应最大速率(v
max
)。实验结果如图1和表1所示。
[0097]
表1腔肠素
‑
h的氘代物与rluc作用的生物发光特性
[0098][0099]
a
最大生物发光强度的波长使用酶标仪测定;
[0100]
b
米氏常数k
m
和v
max
值根据酶水平的小动物活体成像实验,使用grappad prism软件处理所得;
[0101]
c
以腔肠素
‑
h(10μl)的测试结果为基准,折算所有化合物的生物发光强度值。
[0102]
实验结果表明,腔肠素
‑
h的氘代物都具有很强的生物发光强度,并且对比于腔肠素
‑
h,c1和c3的的生物发光强度优于腔肠素
‑
h,c1、c2、c3的米氏常数较腔肠素
‑
h小,特别是147
‑
57的米氏常数约为腔肠素
‑
h的1/3,说明腔肠素
‑
h的氘代设计有助于增强与rluc的亲和力。
[0103]
实施例5:基于腔肠素
‑
h的氘代物在稳定表达rluc的细胞es
‑
2的活性研究
[0104]
5mm氘代腔肠素
‑
h类化合物浓储(现配现用)使用生理盐水(0.9%的氯化钠溶液)稀释为25、10、5、2、1、0.5、0.25、0μm的化合物梯度。将处于对数期的es
‑
2细胞,去除培养基,加入1ml胰蛋白酶,在37℃,5%co2恒温培养箱中消化2min,于显微镜下观察至细胞变圆脱落,加入2ml培养基终止消化,吹散细胞,转移至离心管,离心(1200rpm,6min),去除上清液,加入5ml培养基吹打细胞至单个细胞,使用计数板进行细胞计数,调至4
×
106个/ml,使用排枪(多道移液枪)加至96孔板中,每孔100μl。在37℃,5%co2恒温培养箱中,培养24小时。使用吸痰器去除培养基,使用排枪加入100μl各浓度梯度的化合物,立即成像,曝光时间1s,binning为4,field of view为12.5,f
‑
stop为1,每1min成像一次,连续成像20min。平行实验三次。使用小动物活体成像软件(li4.5.5.19626)进行处理,使用graphpad prism软件对数据,进行处理。
[0105]
实验结果如图2所示,腔肠素
‑
h的氘代物的生物发光强度随化合物的浓度而变化,化合物浓度越大,生物发光强度越强;腔肠素
‑
h的氘代物c1、c2、c3的生物发光强度优于腔肠素
‑
h。
[0106]
实施例6:基于腔肠素
‑
h的氘代化合物在动物水平的生物发光评价
[0107]
将处于对数期的es
‑
2细胞,去除培养基,加入胰蛋白酶,在37℃,5%co2恒温培养箱中消化,于显微镜下观察至细胞变圆脱落,加入培养基终止消化,吹散细胞,转移至离心管,离心,去除上清液,加入无菌生理盐水,洗涤细胞三次,加入无菌生理盐水,吹打细胞,计数,将细胞稀释为1
×
107个/ml。所用的裸鼠为4周大的雌性裸鼠,使用1ml的无菌注射器将
稀释好的细胞注射至裸鼠的右侧腋下,每只裸鼠100μl。在恒温的26℃,保持食物、水、生长环境为无菌且充足的情况下,饲养小鼠4周左右,用小动物活体成像仪进行皮下荷瘤生物发光成像测量。
[0108]
成像结果如图3所示,结果表明,在成像初期,化合物c1、c2和c3与腔肠素
‑
h的活性几乎相当;后续的成像过程中,c1、c2和c3的生物发光强度都比腔肠素
‑
h的强。
[0109]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。