1.本发明涉及可溶性肿瘤坏死因子受体的吸附材料。
背景技术:
2.肿瘤坏死因子(tumor necrosis factor)是存在于血液内的细胞因子的一种,是发挥细胞障碍、抗病毒活性等免疫上的重要作用的物质。已知该肿瘤坏死因子的受体即肿瘤坏死因子受体通常存在于细胞膜中,已知肿瘤坏死因子受体的细胞外区域如果被tumor necrosis factor
‑
α convertase通过酶反应切断,则可溶性肿瘤坏死因子受体1(以下称为stnfr1)、可溶性肿瘤坏死因子受体2(以下称为stnfr2)等的可溶性肿瘤坏死因子受体游离于血液中、尿中。
3.已知可溶性肿瘤坏死因子受体与肿瘤坏死因子结合,抑制其活性。另外,已知可溶性肿瘤坏死因子一般在炎症、败血症、急性呼吸窘迫综合征、c型肝炎、癌、白血病、心房纤颤、心力衰竭、恶液质、自身免疫性疾病等疾病中为在血液中出现的生物标志物的之一,对其生理活性等进行了研究。
4.例如,非专利文献1中报告了,可溶性肿瘤坏死因子受体与肿瘤坏死因子结合,抑制肿瘤坏死因子活性,因此如果能够除去在血液中游离的可溶性肿瘤坏死因子受体,有可能提高源自肿瘤坏死因子的抗肿瘤活性、抗微生物活性。
5.专利文献1中公开了由患者(例如癌患者)的血液等除去stnfr1、stnfr2和可溶性白细胞介素2受体(以下称为sil2r)的方法和系统,为了吸附这些,公开了将stnfr1、stnfr2和sil2r的抗体等固定得到的材料。
6.专利文献2中公开了在水不溶性载体上固定logp(p为辛醇
‑
水系统的分配系数)值为2.5以上的化合物(例如十六烷基胺等)得到的可溶性肿瘤坏死因子受体的吸附材料。
7.另一方面,专利文献3中公开了吸附器,其特征在于,内装有由血液分散液吸附或除去白血球和细胞因子的水不溶性材料。该材料优选孔径为1μm以上100μm以下的孔的孔容积率小于30%,键合亲水性胺残基,并且水不溶性材料的纤维直径为5μm以上20μm以下。
8.专利文献4中公开了一种血液处理用的吸附材料,其是血液的润湿性优异,且血液或血浆的处理时不发生激肽的上升的血液处理用吸附材料,包含排除极限分子量为50,000以上10,000,000以下的高分子的水不溶性载体表面具有官能团,所述官能团具有与不需要的物质的结合性,表面的总带电量为
‑
30μeq/g以上。
9.专利文献5中公开了吸附血液中的粒细胞、单核细胞和细胞因子的吸附材料。该材料表面的zeta电位优选为
‑
20mv以上10mv以下,吸附材料为纤维形状的情况下,从实用的角度考虑,其纤维直径优选为4μm以上20μm以下。
10.专利文献6公开了通过规定配体中含有的氨基和酰胺基的导入量,吸附细胞因子和活化白血球
‑
活化血小板复合体的吸附材料。
11.专利文献7中公开了通过规定材料表面的展开长度比、粗糙度,吸附活化白血球
‑
活化血小板复合体的吸附材料。
12.专利文献8中公开了通过将纤维直径和多孔性最优化,抑制吸附柱的压力损失的吸附材料。
13.现有技术文献专利文献专利文献1:日本特表2008
‑
511340号公报专利文献2:日本特开2009
‑
178523号公报专利文献3:日本特开2007
‑
202634号公报专利文献4:日本特开平6
‑
007430号公报专利文献5:日本特开2006
‑
312804号公报专利文献6:国际公开第2018/047929号专利文献7:国际公开第2018/225764号专利文献8:国际公开第2020/026698号非专利文献非专利文献1:笠仓新平等人,
サイトカイン・ケモカインの
全
て
,日本医学馆,2004年,第三版,p.279
‑
298非专利文献2:tosoh separation report n0.113,p.1非专利文献3:吉田文武等人,化学工学
と
人工臓器,共立出版,1997年,第二版,p.187。
技术实现要素:
14.发明要解决的课题专利文献2中,从吸附性能、选择性的角度考虑,优选水不溶性载体为水不溶性多孔质载体,使水不溶性多孔质载体的排除极限分子量为20000da以上且小于100000da,作为上限的100000da如果用下面的方法换算,则为半径0.5nm左右的微孔,估计可溶性肿瘤坏死因子受体的半径为2nm左右,因此认为作为可溶性肿瘤坏死因子受体的吸附材料是不合适的。在此,排除极限分子量100000da的材料所具有的微孔半径根据与抗体结合的材料表面上具有的微孔孔径和排除极限分子量的关系换算(非专利文献2)。对于可溶性肿瘤坏死因子受体的半径,由于可溶性肿瘤坏死因子受体为约20000da,因此根据其分子量和斯托克斯半径的关系换算(非专利文献3)。
15.另外,专利文献2中记载具有多孔结构的水不溶性载体与表面多孔性相比更优选整体多孔性,整体多孔性的情况下,有可能诱发由于材料的拉伸强度降低导致的材料自身的断裂,因此,从安全性的角度考虑品质管理有可能变得繁杂。要说明的是,作为固定于载体的化合物,实施例中仅示出了正十六烷基胺(ch3(ch2) 15
nh2;σf=7.22),对于多胺衍生物没有任何公开。另外,对于材料表面附近的电荷等没有任何公开或提示。
16.专利文献3中记载了孔是为了形成血球通过的流路而贯通材料的物质,包含纤维与纤维之间的间隙等。即,没有提及在材料表面形成的微孔。并且,作为其孔径,优选1μm以上100μm以下的孔的孔容积率小于30%,且比100μm以上的孔的存在量多,对于在材料表面形成的纳米级别的微孔没有提及。要说明的是,作为细胞因子之一,例示了肿瘤坏死因子
‑
α,但是对于与细胞因子、血球成分不同的可溶性肿瘤坏死因子受体没有任何公开或提示。
17.专利文献4中,从与血浆蛋白质、血浆中的各种成分的结合性的角度规定排除极限分子量,从控制激肽的生成性的角度考虑规定表面的总带电量,从而进行材料设计,对于可溶性肿瘤坏死因子受体、该受体与材料表面的状态的关系没有任何公开或提示。
18.专利文献5中对于与细胞因子、血球成分不同的可溶性肿瘤坏死因子受体、该受体与材料表面的状态的关系没有任何公开或提示。要说明的是,实施例中公开如果吸附材料的纤维直径达到10μm~17μm,嗜中性粒细胞、单核细胞等的血球成分的吸附率大大降低,因此认为使用专利文献5记载的吸附材料除去细胞因子与血球成分的情况下,吸附材料的纤维直径为几μm程度是合适的。
19.专利文献6~8中对于可溶性肿瘤坏死因子受体、该受体与材料表面的状态的关系没有任何公开或提示,也没有进行适合可溶性肿瘤坏死因子受体的吸附的材料设计。
20.为此,本发明的目的在于,提供高效率地吸附可溶性肿瘤坏死因子受体的材料。
21.用于解决课题的手段本发明人等认为为了提高可溶性肿瘤坏死因子受体的吸附效率,根据可溶性肿瘤坏死因子受体的特性设计材料表面的状态是重要的,进行了深入研究,结果发现材料表面的状态中,材料表面的微孔的峰半径和zeta电位是重要的,通过将两者控制为适当的范围,可有效吸附可溶性肿瘤坏死因子受体,从而完成了本发明。
22.即,本发明包含以下的[1]~[8]。
[0023]
[1] 可溶性肿瘤坏死因子受体的吸附材料,其含有表面多孔性的水不溶性高分子材料,由通过差示扫描量热仪测定的熔点的分布求出的上述水不溶性高分子材料的表面的微孔孔径分布曲线中,峰半径为1~80nm,上述水不溶性高分子材料的ph7.4下的zeta电位为
‑
15~15mv,上述水不溶性高分子材料的形状为纤维、颗粒或膜。
[0024]
[2] 根据上述[1]所述的吸附材料,其中,上述水不溶性高分子材料的形状是纤维直径为25~65μm的纤维或颗粒直径为25~65μm的颗粒。
[0025]
[3] 根据上述[1]或[2]所述的吸附材料,其中,在包含上述水不溶性高分子材料的中心的垂直于长度方向的截面中,以非多孔部分的面积为a1、多孔部分的面积为a2的情况下,由下式(1)求出的非多孔部分的面积的比例b为10%以上且90%以下,b=a1/(a1 a2)
×
100
ꢀ・・・
式(1) 。
[0026]
[4] 根据上述[1]~[3]中任一项所述的吸附材料,其中,上述峰半径为1~30nm。
[0027]
[5] 根据上述[1]~[4]中任一项所述的吸附材料,其中,上述水不溶性高分子材料的微孔体积为0.6~1.2cm3/g。
[0028]
[6] 根据上述[1]~[5]中任一项所述的吸附材料,其中,上述水不溶性高分子材料的表面上键合有含有氨基的配体。
[0029]
[7] 根据上述[1]~[6]中任一项所述的吸附材料,其中,上述水不溶性高分子材料含有聚苯乙烯、聚乙烯或聚丙烯作为主成分。
[0030]
[8] 吸附柱,其具有上述[1]~[7]中任一项所述的吸附材料。
[0031]
发明效果本发明的吸附材料可高效率地除去可溶性肿瘤坏死因子受体,因此作为体外循环
用的载体是有用的。
附图说明
[0032]
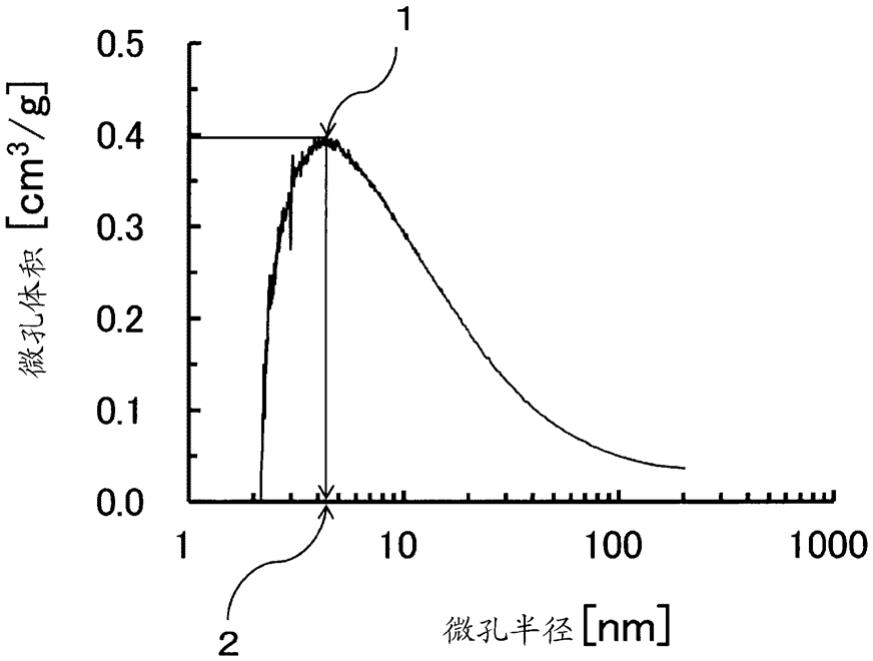
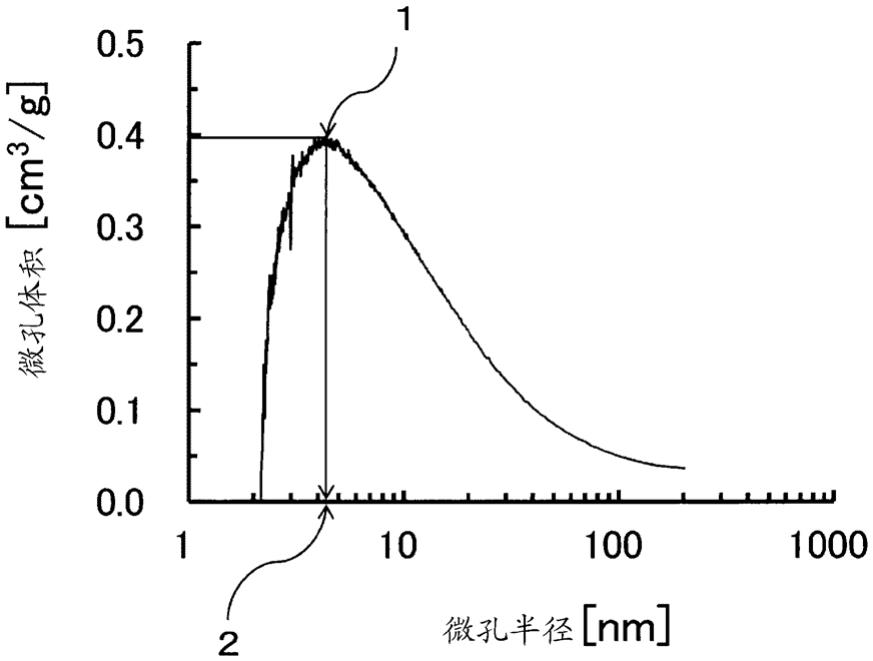
[图1] 用于说明微孔孔径分布曲线中的峰半径的图。
[0033]
[图2] 对于本发明的吸附材料的一例,包括中心的垂直于长度方向切断得到的截面图(纤维形状)。
[0034]
[图3] 对于本发明的吸附材料的一例,包括中心的垂直于长度方向切断得到的截面图(圆球()颗粒形状)。
[0035]
[图4] 对于本发明的吸附材料的一例,包括中心的垂直于长度方向切断得到的截面图(椭圆体颗粒形状)。
[0036]
[图5] 对于本发明的吸附材料的一例,包括中心的垂直于长度方向切断得到的截面图(膜形状)。
[0037]
[图6] 对于本发明的吸附材料的一例,用于说明包括中心的垂直于长度方向切断得到的截面图(纤维形状或颗粒形状)中的多孔部分的面积a2与非多孔部分的面积a1的图。
[0038]
[图7] 对于本发明的吸附材料的一例,用于说明包括中心的垂直于长度方向切断得到的截面图(膜形状)中的多孔部分的面积a2与非多孔部分的面积a1的图。
具体实施方式
[0039]
以下详细说明本发明。
[0040]
本发明的可溶性肿瘤坏死因子受体的吸附材料的特征在于,含有表面多孔性的水不溶性高分子材料,在由通过差示扫描量热仪测定的熔点分布求出的上述水不溶性高分子材料的表面的微孔孔径分布曲线中,峰半径为1~80nm,上述水不溶性高分子材料的ph7.4下的zeta电位为
‑
15~15mv,上述水不溶性高分子材料的形状为纤维、颗粒或膜。
[0041]
"可溶性肿瘤坏死因子受体"是指可溶化(soluble)的,在血液中、尿中游离的肿瘤坏死因子(tumor necrosis factor)的受体,作为可溶性肿瘤坏死因子受体(stnfr),是指可溶性肿瘤坏死因子受体1(stnfr1)、可溶性肿瘤坏死因子受体2(stnfr2)的2种。本发明的吸附材料是吸附stnfr1、stnfr2两者的材料。
[0042]“吸附材料”是指具有可吸附特定的物质、且不容易剥离的性质的材料。吸附的原理没有特别限定,是指通过例如静电相互作用、疏水性相互作用、氢键、范德华力等分子间力进行吸附的材料。
[0043]“表面多孔性”是指微孔存在于材料的表面,微孔不贯通材料的状态。认为表面多孔性的材料与材料的整体具有微孔的整体多孔性材料相比,在可快速达成物质移动、保持强度方面优异。
[0044]“水不溶性高分子材料”是不溶于水的高分子材料。在此,不溶于水是指将水不溶性高分子材料加入水中前后的干燥重量变化为1重量%以下。该干燥重量变化是:将水不溶性高分子材料浸渍于水不溶性高分子材料的干燥重量的9倍量的37℃的水中1小时后,将该水不溶性高分子材料用镊子等提起,将水不溶性高分子材料在50℃以下真空干燥至恒量后所残留的固体成分的干燥重量相对于浸渍前的水不溶性高分子材料的干燥重量的比例。没有进行在水中的不溶化的高分子材料在实际使用的情况下有来自高分子材料的溶出物增
多的危险性,在安全上不优选。
[0045]
作为水不溶性高分子材料的材质,如果是水不溶性的、是高分子,则对其化学结构、物理结构没有特别限定,可列举例如在重复结构中含有芳基、羟基等具有与碳阳离子的反应性的官能团的高分子材料,例如聚(芳香族乙烯基化合物)、聚酯、聚砜、聚醚砜、聚苯乙烯或聚乙烯醇等的合成高分子或纤维素、胶原蛋白、壳多糖、壳聚糖或葡聚糖等的天然高分子。这些高分子可以使用均聚物、共聚物、掺混或合金化使用。特别是血液净化用途中,优选含有作为不具有羟基的高分子材料的选自聚(芳香族乙烯基化合物)、聚对苯二甲酸乙二醇酯、聚对苯二甲酸丁二醇酯、聚苯乙烯、聚砜和聚醚砜中的1种以上聚合物,更优选含有聚苯乙烯、聚乙烯或聚丙烯作为主成分。其中,单位重量的芳香环的数量多,容易通过傅克反应等导入各种官能团、反应性官能团,因此特别优选含有聚苯乙烯作为主成分。在此,主成分是指构成水不溶性高分子材料的成分中含量最多的成分。水不溶性高分子材料为海岛型复合纤维形状的情况下,可列举例如海成分为聚苯乙烯或将聚苯乙烯与聚丙烯混炼得到的合金,岛成分含有聚丙烯或聚乙烯的任意一种以上的复合纤维。要说明的是,这些水不溶性高分子材料通常可购入或通过公知的方法制造。
[0046]
另外,水不溶性高分子材料不仅包括进行表面处理前的水不溶性高分子材料,还包括进行了表面处理的水不溶性高分子材料,例如实施了配体的导入反应等后的水不溶性高分子材料。
[0047]“差示扫描量热仪”被称为differential scanning calorimetry(以下也称为dsc),近年来用作测定湿润状态的样品的微孔分布的装置。通过dsc测定微孔的原理是由于封闭在纳米尺寸的微孔中的冰的熔点比通常的大块冰(熔点:0℃)低,因此利用该现象可由dsc曲线的熔点分布显示微孔孔径分布。具体而言,微孔孔径分布可由dsc曲线的熔点分布按照ishikiriyama等人的方法(journal of colloid and interface science,1995年,171卷,p.92
‑
102)算出。要说明的是,样品中含有水溶性的杂质等的情况下,会产生杂质导致的水的熔点下降,因此有时不可适用微孔孔径分布分析,因此需要在没有水溶性的杂质的状态下供于测定。dsc的测定条件如下。
[0048]
dsc装置:ta instruments 公司制 dsc q100测定温度范围:约
‑
55℃~5℃升温速度:0.3℃/min样品量:约6mg(dsc曲线规格化为5mg进行图示)样品容器:铝制密闭型样品容器温度
・
热量校正:纯水(熔点 0.0℃,熔化热 79.7cal/g)样品的前处理: 除去过量的表面附着水(大体积的水)微孔孔径分布曲线中的峰半径如图1所示,是指将通过上述dsc测定的微孔孔径分布(横轴:微孔半径、竖轴:水不溶性高分子材料的每1g湿润重量的微孔体积)作图得到的微孔孔径分布曲线中,水不溶性高分子材料的每1g湿润重量的微孔体积显示最大值的微孔的半径。
[0049]
为了提高stnfr的吸附效率,重要的是相对于stnfr的大小适当控制水不溶性高分子材料的表面上形成的微孔孔径。为此,基于如果stnfr高效地内包于水不溶性高分子材料的微孔内,则吸附效率提高的想法进行了研究,结果发现通过在微孔的表面高频率地形成
适当大小的微孔,可提高stnfr的吸附效率。
[0050]
水不溶性高分子材料的表面的微孔的峰半径由stnfr的吸附量和侵入的容易性考虑,需要为1nm以上,从实现stnfr的选择性吸附的角度考虑,需要为80nm以下。即,水不溶性高分子材料的表面的微孔的峰半径需要为1~80nm,优选为1~30nm,更优选为1~20nm,进一步优选为3~20nm。
[0051]
水不溶性高分子材料表面的微孔例如可通过将水不溶性高分子材料的表面亲水化,使其膨润而形成。例如通过在水不溶性高分子材料的表面固定含有酸性官能团或碱性官能团的配体,将表面亲水化,使其膨润,可在表面形成微孔。在此,作为在水不溶性高分子材料的表面上固定上述配体的反应中使用的溶剂,适当使用硝基苯。进一步地,通过在将水不溶性高分子材料的表面亲水化、使其膨润后将该水不溶性高分子材料浸渍于醇中,也可在表面形成微孔。在此所述的醇可列举甲醇、乙醇、异丙醇等。另外,从也可通过醇浸渍时的温度控制微孔的大小,提高吸附能力的角度考虑,优选为20~60℃,更优选30~50℃。另外,从通过浸渍于醇中的时间也可控制微孔的大小,提高吸附能力的角度考虑,优选5~60分钟,更优选20~40分钟。要说明的是,前述的专利文献6~8的实施例中,没有记载制作氯乙酰胺甲基化针织物时的甲醇的浸渍温度、浸渍时间。
[0052]
水不溶性高分子材料的表面的微孔的峰半径可通过水不溶性高分子材料的聚合物种类、多种聚合物的掺混物或合金化的条件控制。例如通过改变聚苯乙烯与聚丙烯的合金的比率,可控制上述微孔的峰半径。
[0053]“zeta电位”是指双电层中的滑动面与充分远离界面的部分之间的电位差。本技术中的zeta电位是指在ph7.4、温度20
±
5℃的条件下使用电泳法测定的值。
[0054]
没有进行表面处理的聚苯乙烯、聚乙烯、聚丙烯等的水不溶性高分子材料作为zeta电位具有负值,大概为
‑
30mv左右。作为将zeta电位控制为
‑
15mv以上的方法之一,可列举在水不溶性高分子材料的表面固定含有特定的官能团(例如酸性官能团或碱性官能团)的配体的方法。研究的结果,如果使zeta电位为
‑
15mv以上,发现stnfr与水不溶性高分子材料表面的静电相互作用提高,而如果使zeta电位为15mv以上,则有可能对红血球产生不良影响,因此zeta 电位需要为
‑
15~15mv,优选为
‑
10~12mv,更优选为
‑
4~10mv。上述的峰半径与zeta电位的优选方式可任意组合。
[0055]
作为水不溶性高分子材料的形状,适当使用纤维、颗粒或膜。考虑到用作吸附材料,优选比表面积大,柔软的可变形的操作性优异的纤维形状、特别是海岛型复合纤维形状。其中,优选海成分为聚苯乙烯、岛成分为聚丙烯的海岛型复合纤维形状。另外考虑到使用时的吸附材料的填充、液体的流路的均匀性,纤维形状中,优选机织物、无纺布、针织物形状。可溶性肿瘤坏死因子受体的吸附材料至少一部分含有水不溶性高分子材料即可,可以为水不溶性高分子材料单独,也可以为将适当的增强材料固定或混合于水不溶性高分子材料得到的材料。固定化或混合操作可以在加工成形状前进行,也可以在加工后进行。
[0056]“纤维直径”是指随机采取形成构成水不溶性高分子材料的机织物、无纺布或针织物的纤维的小片样品10个,使用扫描型电子显微镜分别拍摄照片,每张照片测定10处(共计100处)的纤维直径得到的值的平均值。此时的观察倍率是使纤维直径为照片长边的长度的30~80%的倍率。另外,是多个纤维成束的复丝的情况下,将构成复丝的单丝的直径作为纤维直径。
[0057]
水不溶性高分子材料的形状为纤维的情况下,作为该纤维的纤维直径,从抑制止血所需要的血小板的吸附的角度考虑,优选为25μm以上,更优选为30μm以上。另外,从为了吸附stnfr而确保纤维的比表面积的角度考虑,优选为65μm以下,更优选为50μm以下。即,作为该纤维的纤维直径,优选25~65μm,更优选30~50μm。任意的优选下限值可与任意的优选上限值组合。
[0058]“颗粒直径”是指随机采取形成构成水不溶性高分子材料的颗粒的珠等的小片样品10个,使用扫描型电子显微镜分别拍摄照片,每张照片测定10处(共计100处)的颗粒的直径得到的值的平均值。此时的观察倍率为使颗粒直径为照片长边的长度的30~80%的倍率。
[0059]
水不溶性高分子材料的形状为颗粒的情况下,作为该颗粒的颗粒直径,从抑制血小板的吸附的角度考虑,优选25μm以上,更优选30μm以上。另外,从为了stnfr的吸附而确保颗粒的比表面积的角度考虑,优选65μm以下,更优选50μm以下。即,作为该颗粒的颗粒直径,优选25~65μm,更优选30~50μm。任意的优选下限值可与任意的优选上限值组合。另外,基于不引起压力损失的增大等的理由,优选粒径分布窄。
[0060]
在包括水不溶性高分子材料的中心的垂直于长度方向的方向的截面(图2~7)中,以非多孔部分的面积为a1、多孔部分的面积为a2的情况下,可按照下式(1)求出非多孔部分的面积的比例b。该方法中,将截面通过四氧化钌染色冷冻超薄切片法进行前处理,通过透射型电子显微镜em
‑
1400plus(日本电子公司制)在加速电压100kv的条件下以8000倍的倍率随机观察包括水不溶性高分子材料的中心的垂直于长度方向的截面,拍摄截面图像100处。在拍摄的截面图像中,非多孔部分的面积a1与多孔部分的面积a2的辨别可通过存在于截面图像中的材料表面的微孔判断。非多孔部分的面积a1是指存在小于10nm的微孔的区域的面积和不存在微孔的区域的面积之和,多孔部分的面积a2是存在10nm以上的微孔的区域的面积。要说明的是,截面中配置2种成分以上的水不溶性高分子材料的情况下的非多孔部分的面积a1与多孔部分的面积a2的边界可使用截面上配置的各成分(例如海成分与岛成分)的边界。该情况下,将存在小于10nm的微孔的成分的面积和不存在微孔的成分的面积之和作为非多孔部分的面积a1,将存在10nm以上的微孔的成分的面积作为多孔部分的面积a2而算出。面积的算出可使用图像编辑软件(photoshop elements.14.0 adobe公司制)。
[0061]
图2是显示水不溶性高分子材料为纤维形状的情况下的包括中心的垂直于长度方向的截面的图的例子。中心为垂直于长度方向的方向的截面上的2根中心线(7)相交的点(4)。图3是圆球的颗粒形状、图4是椭圆体的颗粒形状、图5是膜形状,与图2同样,是显示包括中心的垂直于长度方向的方向的截面的图的例子。要说明的是,如图3那样颗粒是圆球的情况下,没有长度方向的概念,因此是包括中心的截面即可。得到的截面的图像分析的一例示于图6或图7。以微孔孔径小于10nm的非多孔部分(12)(包括不存在微孔的区域)的面积之和为a1,以微孔孔径10nm以上的多孔部分(11)的面积之和为a2,可求出以下的式(1)所示的非多孔部分的面积的比例b。
[0062]
b=a1/(a1 a2)
×
100
ꢀ・・・
式(1)上述截面中,非多孔部分的面积的比例b如果过小,则材料的强度降低,作为吸附材料不能保持形态,从该角度考虑,优选非多孔部分的面积的比例b为10%以上,而从如果非多孔部分的面积的比例b过大,则不能发挥吸附性能的角度考虑,优选为90%以下。即,优选非多孔部分的面积的比例b为10~90%,更优选为20~80%,进一步优选为30~70%。任意的优选
下限值可与任意的优选上限值组合。
[0063]“微孔体积”是由通过dsc测定的熔点分布求出的每1g水不溶性高分子材料的微孔体积。利用dsc的微孔体积的测定方法与上述测定微孔的方法同样,由dsc曲线的熔点分布按照ishikiriyama等人的方法(journal of colloid and interface science,1995年,171卷,p.92
‑
102)可算出微孔体积。
[0064]
如果水不溶性高分子材料的微孔体积过小,则不能充分提高吸附性能,因此优选该微孔体积为0.06cm3/g以上,另一方面,如果上述微孔体积过大,则得不到stnfr的选择性吸附特性,从该角度考虑优选为1.2cm3/g以下。即,水不溶性高分子材料的微孔体积优选为0.06~1.2cm3/g,更优选为0.3~1.2cm3/g。进一步地,通过使水不溶性高分子材料的微孔体积为0.6~1.2cm3/g,可期待体外循环开始2小时后可溶性肿瘤坏死因子受体的吸附率达到80%以上,认为该情况下,能够在可定期到医院治疗的4小时以内结束处置,因此可减轻患者的负担。在此,对于上述水不溶性高分子材料的微孔体积与上述水不溶性高分子材料的纤维直径或颗粒直径,优选的方式可任意组合。
[0065]
上述的水不溶性高分子材料的表面上可以键合配体。本说明书中,“配体”是指键合在水不溶性高分子材料的表面的化合物,如果含有酸性官能团或碱性官能团,则对其化学结构没有特别限制,可列举例如含有作为酸性官能团(阴离子性官能团)的磺酸基或羧基的化合物或含有作为碱性官能团(阳离子性官能团)的氨基的化合物。本实施方式中,作为配体,优选含有碱性官能团的化合物,特别优选含有氨基的化合物。要说明的是,上述官能团可以组合多种相同或不同的官能团。要说明的是,配体如果具有上述酸性官能团或碱性官能团,则可以进一步具有中性官能团,作为该中性官能团,例如甲基或乙基等的烷基或苯基、烷基取代的苯基(例如对(p)
‑
甲基苯基、间(m)
‑
甲基苯基、邻(o)
‑
甲基苯基、对(p)
‑
乙基苯基、间(m)
‑
乙基苯基或邻(o)
‑
乙基苯基等)或卤素原子取代的苯基(例如对(p)
‑
氟苯基、间(m)
‑
氟苯基、邻(o)
‑
氟苯基、对(p)
‑
氯苯基、间(m)
‑
氯苯基或邻(o)
‑
氯苯基等)等的芳基键合于含有酸性官能团或碱性官能团的化合物上得到的化合物(例:键合了对(p)
‑
氯苯基的四亚乙基五胺)包括在配体中。此时,中性官能团与配体可以直接键合,也可以通过间隔基团键合(参与该键合的间隔基团称为间隔基团1)。作为该间隔基团1,可列举例如尿素键、酰胺键、氨基甲酸酯键。
[0066]“氨基”可列举例如源自甲基胺、乙基胺、丙基胺、丁基胺、戊基胺、己基胺、庚基胺、辛基胺或十二烷基胺等的伯胺的氨基、源自甲基己基胺、二苯基甲基胺、二甲基胺等的仲胺的氨基、源自烯丙基胺等的具有不饱和烷基链的胺的氨基、源自三甲基胺、三乙基胺、二甲基乙基胺、苯基二甲基胺、二甲基己基胺等的叔胺的氨基、源自1
‑
(3
‑
氨基丙基)咪唑、吡啶
‑2‑
胺、3
‑
磺基苯胺等的具有芳香环的胺的氨基或源自三(2
‑
氨基乙基)胺、乙二胺、二亚乙基三胺、三亚乙基四胺、四亚乙基五胺、二亚丙基三胺、聚乙烯亚胺、n
‑
甲基
‑
2,2
’‑
二氨基二乙基胺、n
‑
乙酰基乙二胺、1,2
‑
双(2
‑
氨基乙氧基乙烷)等的通过烷基链、芳香族化合物、杂环化合物、单环化合物等键合2个以上氨基的化合物(以下称为“多胺”)的氨基,优选为源自多胺的氨基,特别是优选为源自乙二胺、二亚乙基三胺、三亚乙基四胺或四亚乙基五胺的氨基,更优选源自四亚乙基五胺的氨基。另外,氨基更优选源自伯胺或仲胺的氨基。另外,优选logp(p为辛醇
‑
水系的分配系数)值小于2.5的氨基,例如也适合使用四亚乙基五胺。
[0067]
水不溶性高分子材料和含有酸性官能团或碱性官能团的配体可以直接键合,在上
述水不溶性高分子材料与上述配体之间也可以存在源自反应性官能团的间隔基团(参与该键合的间隔基团也称为间隔基团2)。作为该间隔基团2,具有尿素键、酰胺键、醚键、酯键、氨基甲酸酯键等的电中性的化学键即可,优选具有酰胺键或尿素键。
[0068]
作为促成上述水不溶性高分子材料与上述配体的键合的反应性官能团,可列举例如卤代乙酰基、卤代乙酰胺基甲基或卤代烷基等的活性卤素基、环氧基、羧基、异氰酸基、异硫氰酸基或酸酐基,从具有适度的反应性的角度考虑,优选活性卤素基,更优选卤代乙酰胺基甲基。作为导入了反应性官能团的水不溶性高分子材料的具体例子,可列举加成了氯乙酰胺基甲基的聚苯乙烯、加成了氯乙酰胺基甲基的聚砜。
[0069]
反应性官能团可通过预先使水不溶性高分子材料与适当的试剂反应键合于水不溶性高分子材料。例如水不溶性高分子材料为聚苯乙烯、反应性官能团为氯乙酰胺基甲基的情况下,通过使聚苯乙烯与n
‑
羟甲基
‑
α
‑
氯乙酰胺反应,可得到键合了氯乙酰胺基甲基的聚苯乙烯。对于键合了氯乙酰胺基甲基的聚苯乙烯,通过例如与具有氨基的四亚乙基五胺反应,得到通过乙酰胺基甲基键合了四亚乙基五胺的聚苯乙烯。该情况下,乙酰胺基甲基相当于间隔基团2,四亚乙基五胺相当于配体。另外,对于键合了氯乙酰胺基甲基的聚苯乙烯,与例如具有氨基的四亚乙基五胺反应,进而与氯苯基异氰酸酯反应,得到通过尿素键键合了氯苯基的四亚乙基五胺通过乙酰胺基甲基键合的聚苯乙烯。水不溶性高分子材料的材质、间隔基团(间隔基团1和间隔基团2)、配体可任意组合。作为键合了配体的海成分的例子,可列举含有源自乙二胺、二亚乙基三胺、三亚乙基四胺或四亚乙基五胺的氨基的化合物通过乙酰胺基甲基键合的聚苯乙烯、含有源自乙二胺、二亚乙基三胺、三亚乙基四胺或四亚乙基五胺的氨基的化合物通过乙酰胺基甲基键合的聚砜、含有源自乙二胺、二亚乙基三胺、三亚乙基四胺或四亚乙基五胺的氨基的化合物通过乙酰胺基甲基键合的聚醚砜。
[0070]
酸性官能团或碱性官能团的含量没有特别限定,如果其含量过少,则不能充分提高对血液成分等的具有电荷的stnfr的吸附性能。而如果其含量过多,则认为亲水性提高,水不溶性高分子材料的强度降低,因此作为每1g水不溶性高分子材料的干燥重量的酸性官能团或碱性官能团的含量,例如为0.5~5.0mmol。
[0071]
上述酸性官能团或碱性官能团的含量可通过使用盐酸或氢氧化钠水溶液的酸碱滴定法测定。
[0072]
从提高与作为吸附对象的stnfr的相互作用的角度考虑,优选含有酸性官能团或碱性官能团的配体键合于水不溶性高分子材料的表面(特别是,海岛型复合纤维形状的情况下为表面的海成分)。
[0073]
本实施方式的可溶性肿瘤坏死因子受体的吸附材料例如可用作体外循环用的吸附柱的载体、血液制剂制造用的柱的载体、stnfr的分离、纯化柱的载体。作为可将上述吸附材料用作体外循环用的吸附柱的载体的患者,如果是血液中高浓度游离stnfr1、stnfr2的疾病的患者,则没有特别限定,例如可通常用于一般炎症、败血症、急性呼吸窘迫综合征、c型肝炎、癌、白血病、心房纤颤、心力衰竭、恶液质、自身免疫性疾病等的患者。其中,可列举败血症、急性呼吸窘迫综合征、癌等的患者。
[0074]
特别是,癌患者由于放射线治疗、抗癌剂的副作用免疫被抑制,因此由癌患者高效率地吸附抑制免疫的可溶性肿瘤坏死因子受体的材料可期待改善癌患者的qol。癌中,可适用于白血病、甲状腺癌、肝癌、肾癌、肺癌、食道癌、胃癌、胰脏癌、乳癌、前列腺癌、子宫癌、宫
颈癌、膀胱癌、恶性脑肿瘤、咽癌、淋巴结癌等的治疗。
[0075]
另外,上述吸附载体由于其高亲和性可保持可溶性肿瘤坏死因子受体,因此可将可溶性肿瘤坏死因子受体与其它成分分离,与其它成分分离后,将上述吸附载体用规定的溶液冲洗、浸渍,可使保持于吸附载体的可溶性肿瘤坏死因子受体由吸附载体脱离。作为在吸附载体上吸附可溶性肿瘤坏死因子受体的方法,例如可制作将上述的吸附材料填充于如注射筒的容器、或者填充于微型柱、微芯得到的材料,使含有可溶性肿瘤坏死因子受体的液体在它们中通过、或者浸渍来实施。作为使可溶性肿瘤坏死因子受体从吸附载体脱离的方法,例如可在吸附载体中通过调整了离子浓度、盐浓度的溶液。由此,可得到纯度高的可溶性肿瘤坏死因子受体。即,也可用作可溶性肿瘤坏死因子受体的纯化用的材料。要说明的是,本吸附材料可与通过离子、疏水性、分子量、孔径进行分离
・
纯化的其它材料并用进行有效地分离
・
纯化。
[0076]
另外,本发明提供使用上述吸附材料的可溶性肿瘤坏死因子受体的除去方法。
[0077]
将上述的吸附材料用作体外循环用的吸附柱的载体的情况下,作为可溶性肿瘤坏死因子受体的除去方法的方式之一,通过将吸附柱与安装于血液循环装置的血液回路连接,使患者的血液循环,可净化患者的血液,得到治疗效果。此时,静脉血动脉血均可。更具体而言,可列举将患者的血液直接通入吸附柱,除去血液中含有的可溶性肿瘤坏死因子受体的方法(直接血液灌流疗法)、将患者的血液通过血浆分离器分离,将得到的血浆成分通入吸附柱,除去血浆成分中含有的可溶性肿瘤坏死因子受体的方法(血浆灌流疗法)。通过这些方法净化的血液等返回至患者。要说明的是,对于肾衰竭的患者,可将吸附柱与人工肾脏并用。对于肺功能衰竭的患者,可与人工呼吸器、体外式膜型人工肺并用。
[0078]
另外,作为可溶性肿瘤坏死因子受体的除去方法的别的方式,可列举使用上述的吸附材料由血液制剂除去血液制剂制造中产生的可溶性肿瘤坏死因子受体的方法。在此,血液制剂是指全血制剂、浓集红血球制剂、血小板制剂、白蛋白制剂、免疫球蛋白制剂、血液凝固因子制剂或血液凝固因子相关制剂等的输血中使用的各种血液制剂。具体而言,在由通过献血采集的血液制造血液制剂的任意步骤中,填充了上述的吸附材料的吸附柱上连接管、回路或血液制剂袋,使用泵、振荡机使血液、血液制剂的中间制品、血液制剂等通过吸附柱,由此除去血液、血液制剂的中间制品、血液制剂等中含有的可溶性肿瘤坏死因子受体的方法。另外,在将要向患者施与血液制剂前,使血液制剂通过填充了上述的吸附材料的吸附柱也可除去血液制剂中含有的可溶性肿瘤坏死因子受体。
[0079]
另外,本发明提供具有上述的吸附材料的吸附柱。
[0080]“吸附柱”是指至少具有液体入口部、壳体部、液体出口部,在壳体部具有本发明的可溶性肿瘤坏死因子受体的吸附材料(以下也称为stnfr吸附材料)的柱。作为柱,可列举例如径向流动型的柱。
[0081]
本实施方式的吸附柱通过使液体通过,可由该液体中吸附stnfr,因此可作用由含有stnfr的液体纯化或除去作为目标的stnfr的用途,例如可用于特定的stnfr的分离等。
[0082]
作为吸附柱的容器形状,为具有含有stnfr的液体(以下称为液体)的入口部和出口部、壳体部的容器、可在该壳体部内填充stnfr吸附材料的形状即可。作为一个实施方式,可列举可在内部填充将stnfr吸附材料卷绕于管、制成圆筒状的物质(以下称为圆筒)的容器,液体由圆筒的外周进入、流向圆筒的内侧后,该液体流出容器外的容器或液体由圆筒的
内侧流入流向圆筒的外侧后该液体流出容器外的容器。从制造效率、抑制处理液的短路的角度考虑,优选对侧面具有孔的管缠绕stnfr吸附材料的结构,具体而言,可列举径向流动型的容器,其具有:在长度方向的侧面具有为了流出所供给的液体而设置的孔的中心管;填充于上述中心管的周围而吸附上述液体中含有的目标物质的stnfr吸附材料;与上述中心管的上游端连通以使流入进来的上述液体通过上述中心管中、以防止上述液体不通过上述中心管而与上述stnfr吸附材料接触的方式配置的板;以封闭上述中心管的下游端、将上述stnfr吸附材料固定于上述中心管周围的空间的方式配置的板,另外,容器的形状可列举圆柱状或三棱柱状、四棱柱状、六棱柱状或八棱柱状等的棱柱状容器,但不限于该结构。另外作为别的实施方式,可考虑内部具有可填充切成圆形的stnfr吸附材料的圆筒状的空间的、具有液体导入口和液体排出口的容器。具体而言,可列举下述容器,其内部具有:具有为了流出所供给的液体而设置的液体导入口的板、具有为了排出所供给的液体而设置的液体排出口的板、填充了切成圆形的stnfr吸附材料的圆筒状的壳体部、具有液体导入口和液体排出口。要说明的是,该情况下,stnfr吸附材料的形状不限于圆形,根据吸附柱的容器形状可适当变更为椭圆形、三角形、四边形等多边形、梯形等任意的形状。
[0083]
作为吸附柱的容器,可列举玻璃制、塑料
・
树脂制、不锈钢制等的容器,容器的尺寸可根据使用目的适当选择,吸附柱的容器的大小等没有特别限制,考虑到临床现场、测定场所的操作性、废弃的容易性,作为材质优选塑料
・
树脂制,大小优选容易手握的大小,整体吸附柱的高度优选为1cm以上30cm以下、外径为1cm以上10cm以下、内容积为200cm3以下。要说明的是,后述实施例中,从测定的简便性考虑,使用填充体积0.94cm3(内径1cm
×
高度1.2cm、外径2cm)的吸附柱,但不限于此。
[0084]
stnfr吸附材料优选层叠于吸附柱内进行填充。在此,层叠是指将stnfr吸附材料2片以上密合重叠,作为层叠填充的方法,例如以成为轴向流动柱的方式将加工为片材形态的stnfr吸附材料多片重叠的方法、以成为径向流动柱的方式将加工为片材形态的stnfr吸附材料卷绕于具有孔的管的方法。
实施例
[0085]
以下,通过实施例具体说明本发明的可溶性肿瘤坏死因子受体的吸附材料,但本发明不限于这些例子。
[0086]
(纤维a的制作)作为海成分使用聚苯乙烯、作为岛成分使用聚丙烯,分别熔融计量,且使其流入组装了每个喷出孔穿设256个岛成分用分配孔的海岛复合喷丝头的纺丝组件,制成海岛复合流,进行熔融喷出。将岛比率控制为50重量%,将水不溶性高分子材料的表面至最外侧岛成分的距离调整为2μm,采取纤维直径为20μm的海岛型复合纤维a(以下称为纤维a)。
[0087]
(纤维b的制作)作为海成分使用聚苯乙烯,作为岛成分使用聚丙烯,分别熔融计量,使其流入组装了每个喷出孔穿设256个岛成分用分配孔的海岛复合喷丝头的纺丝组件,制成海岛复合流,进行熔融喷出。将岛比率控制为50重量%,将水不溶性高分子材料的表面至最外侧岛成分的距离调整为2μm,采取纤维直径为30μm的海岛型复合纤维b(以下称为纤维b)。
[0088]
(纤维c的制作)
作为海成分使用聚苯乙烯,作为岛成分使用聚丙烯,分别熔融计量,使其流入组装了每个喷出孔穿设256个岛成分用分配孔的海岛复合喷丝头的纺丝组件,制成海岛复合流,进行熔融喷出。将岛比率控制为50重量%,将海岛型复合纤维的表面至最外侧岛成分的距离调整为2μm,采取纤维直径为60μm的海岛型复合纤维c(以下称为纤维c)。
[0089]
(纤维d的制作)作为海成分使用聚苯乙烯,作为岛成分使用聚丙烯,分别熔融计量,使其流入组装了每个喷出孔穿设256个岛成分用分配孔的海岛复合喷丝头的纺丝组件,制成海岛复合流,进行熔融喷出。将岛比率控制为50重量%,将海岛型复合纤维的表面至最外侧岛成分的距离调整为2μm,采取纤维直径为70μm的海岛型复合纤维d(以下称为纤维d)。
[0090]
(无纺布e的制作)按照日本特开2006
‑
312804号公报的说明书中记载的方法,使用以下的成分,以800m/分钟的纺丝速度、拉伸倍率为3倍的制丝条件得到36岛的海岛复合纤维,岛进一步为芯鞘复合纤维。
[0091]
岛的芯成分;聚丙烯岛的鞘成分:以聚苯乙烯90重量%、聚丙烯10重量%的比例混炼海成分:以对苯二甲酸乙二醇酯作为主要的重复单元,作为共聚成分含有间苯二甲酸5
‑
磺酸钠3重量%的共聚聚酯复合比率(重量比): 芯:鞘:海=44:44:12以上述的36岛的海岛复合纤维85重量%、作为骨材的直径20μm的聚丙烯15重量%的比例均匀混合,将它们加工为片状。通过将该片进行针刺得到无纺布。接着,通过将该无纺布用90℃的氢氧化钠水溶液处理,采取芯鞘纤维的直径为5μm、松密度为0.05g/cm3(总单位面积重量250g/m2)的无纺布e(以下称为无纺布e)。
[0092]
(针织物a~d的制作)使用纤维a~d,调整圆筒针织机(机种名:圆型针织机 mr
‑
1,丸善产业株式会社)的线圈密度调整刻度,分别制作单位面积重量为0.0039g/cm2、松密度为0.22g/cm3的圆筒针织物a~d(以下称为针织物a~d)。
[0093]
(氯乙酰胺基甲基化针织物的制作)将n
‑
羟甲基
‑
α
‑
氯乙酰胺(以下称为nmca)23g添加于硝基苯310g和98重量%硫酸310g的混合溶液中,在10℃搅拌直至nmca溶解,制成nmca溶液。接着,在硝基苯20g、98重量%硫酸20g中添加多聚甲醛(以下称为pfa)2g,在20℃搅拌直至pfa溶解,制成pfa溶液。将pfa溶液42g冷却至5℃,混合至nmca溶液643g中,搅拌5分钟,添加10g针织物a,浸渗2小时。将浸渗后的针织物a浸渍于10℃以下的硝基苯500ml中使反应停止后,将附着于该针织物的硝基苯在加热至40℃的甲醇中浸渍30分钟,制作氯乙酰胺基甲基化针织物a。除了将针织物a变更为针织物b~d以外,通过与氯乙酰胺基甲基化针织物a的制作方法同样的方法,分别制作氯乙酰胺基甲基化针织物b~d。
[0094]
(氯乙酰胺基甲基化无纺布的制作)在硝基苯600ml与98重量%硫酸390ml的混合溶液中在20℃溶解pfa3g后,冷却至0℃,加入75.9g的n
‑
羟甲基
‑
α
‑
氯乙酰胺,在5℃以下溶解。在该混合液中浸渍5g的无纺布e,在室温静置2小时。然后,取出无纺布,将该无纺布浸渍于大大过量的10℃以下的甲醇中。将
无纺布在甲醇中浸渍后,进行水洗,干燥,制作氯乙酰胺基甲基化无纺布e。
[0095]
(四亚乙基五胺
‑
对氯苯基化针织物的制作)在以四亚乙基五胺(以下称为tepa)的浓度为5mm、三乙基胺的浓度为473mm的方式分别溶解于500ml的二甲基亚砜(以下称为dmso)得到的液体中,浸渍10g的上述氯乙酰胺基甲基化针织物b,在40℃反应3小时。将该针织物用dmso洗涤3次后,浸渍于以对氯苯基异氰酸酯的浓度为20mm的方式溶解于500ml的dmso中得到的液体中,在30℃反应1小时。然后,由反应液取出针织物,浸渍于与反应液相同量的dmso中进行洗涤,接着用甲醇洗涤,接着浸渍于水中洗涤,得到四亚乙基五胺
‑
对氯苯基化针织物(以下称为吸附材料1)。
[0096]
(四亚乙基五胺
‑
对氯苯基化针织物的制作)在以tepa的浓度达到10mm、三乙基胺的浓度达到473mm的方式分别溶解于500ml的dmso中得到的液体中,浸渍10g的上述氯乙酰胺基甲基化针织物b,在40℃反应3小时。将该针织物用dmso洗涤3次后,浸渍于以对氯苯基异氰酸酯的浓度达到20mm的方式溶解于500ml的dmso中得到的液体中,在30℃反应1小时。然后,由反应液取出针织物,浸渍于与反应液相同量的dmso中进行洗涤,接着用甲醇洗涤,接着浸渍于水中进行洗涤,得到四亚乙基五胺
‑
对氯苯基化针织物(以下称为吸附材料2)。
[0097]
(四亚乙基五胺
‑
对氯苯基化针织物的制作)在以tepa的浓度达到20mm、三乙基胺的浓度达到473mm的方式分别溶解于500ml的dmso得到的液体中,浸渍10g的上述氯乙酰胺基甲基化针织物b,在40℃反应3小时。将该针织物用dmso洗涤3次后,浸渍于以对氯苯基异氰酸酯的浓度达到20mm的方式溶解于500ml的dmso中得到的液体,在30℃反应1小时。然后,由反应液取出针织物,浸渍于与反应液相同量的dmso中进行洗涤,接着用甲醇洗涤,接着浸渍于水中进行洗涤,得到四亚乙基五胺
‑
对氯苯基化针织物(以下称为吸附材料3)。
[0098]
(四亚乙基五胺
‑
对氯苯基化针织物的制作)在以tepa的浓度达到30mm、三乙基胺的浓度达到473mm的方式分别溶解于500ml的dmso得到的液体中,浸渍10g的上述氯乙酰胺基甲基化针织物b,在40℃反应3小时。将该针织物用dmso洗涤3次后,浸渍于以对氯苯基异氰酸酯的浓度达到20mm的方式溶解于500ml的dmso中得到的液体,在30℃反应1小时。然后,由反应液取出针织物,浸渍于与反应液相同量的dmso中进行洗涤,接着用甲醇洗涤,接着浸渍于水中进行洗涤,得到四亚乙基五胺
‑
对氯苯基化针织物(以下称为吸附材料4)。
[0099]
(四亚乙基五胺
‑
对氯苯基化针织物的制作)在以tepa的浓度达到40mm、三乙基胺的浓度达到473mm的方式分别溶解于500ml的dmso中得到的液体中,浸渍10g的上述氯乙酰胺基甲基化针织物b,在40℃反应3小时。将该针织物用dmso洗涤3次后,浸渍于以对氯苯基异氰酸酯的浓度达到20mm的方式溶解于500ml的dmso中得到的液体,在30℃反应1小时。然后,由反应液取出针织物,浸渍于与反应液相同量的dmso中进行洗涤,接着用甲醇洗涤,接着浸渍于水中进行洗涤,得到四亚乙基五胺
‑
对氯苯基化针织物(以下称为吸附材料5)。
[0100]
(四亚乙基五胺
‑
对氯苯基化针织物的制作)在以tepa的浓度达到30mm、三乙基胺的浓度达到473mm的方式分别溶解于500ml的dmso中得到的液体中,浸渍10g的上述氯乙酰胺基甲基化针织物c,在40℃反应3小时。将该
针织物用dmso洗涤3次后,浸渍于以对氯苯基异氰酸酯的浓度达到20mm的方式溶解于500ml的dmso中得到的液体,在30℃反应1小时。然后,由反应液取出针织物,浸渍于与反应液相同量的dmso中进行洗涤,接着用甲醇洗涤,接着浸渍于水中进行洗涤,得到四亚乙基五胺
‑
对氯苯基化针织物(以下称为吸附材料6)。
[0101]
(四亚乙基五胺
‑
对氯苯基化针织物的制作)在以tepa的浓度达到2.5mm、三乙基胺的浓度达到473mm的方式分别溶解于500ml的dmso中得到的液体中,浸渍10g的上述氯乙酰胺基甲基化针织物b,在40℃反应3小时。将该针织物用dmso洗涤3次后,浸渍于以对氯苯基异氰酸酯的浓度达到20mm的方式溶解于500ml的dmso中得到的液体,在30℃反应1小时。然后,由反应液取出针织物,浸渍于与反应液相同量的dmso中进行洗涤,接着用甲醇洗涤,接着浸渍于水中进行洗涤,得到四亚乙基五胺
‑
对氯苯基化针织物(以下称为吸附材料7)。
[0102]
(四亚乙基五胺
‑
对氯苯基化针织物的制作)在以tepa的浓度达到50mm、三乙基胺的浓度达到473mm的方式分别溶解于500ml的dmso中得到的液体中,浸渍10g的上述氯乙酰胺基甲基化针织物b,在40℃反应3小时。将该针织物用dmso洗涤3次后,浸渍于以对氯苯基异氰酸酯的浓度达到20mm的方式溶解于500ml的dmso中得到的液体,在30℃反应1小时。然后,由反应液取出针织物,浸渍于与反应液相同量的dmso中进行洗涤,接着用甲醇洗涤,接着浸渍于水中进行洗涤,得到四亚乙基五胺
‑
对氯苯基化针织物(以下称为吸附材料8)。
[0103]
(四亚乙基五胺
‑
对氯苯基化针织物的制作)在以tepa的浓度达到30mm、三乙基胺的浓度达到473mm的方式分别溶解于500ml的dmso中得到的液体中,浸渍10g的上述氯乙酰胺基甲基化针织物a,在40℃反应3小时。将该针织物用dmso洗涤3次后,浸渍于以对氯苯基异氰酸酯的浓度达到20mm的方式溶解于500ml的dmso中得到的液体,在30℃反应1小时。然后,由反应液取出针织物,浸渍于与反应液相同量的dmso中进行洗涤,接着用甲醇洗涤,接着浸渍于水中进行洗涤,得到四亚乙基五胺
‑
对氯苯基化针织物(以下称为吸附材料9)。
[0104]
(四亚乙基五胺
‑
对氯苯基化针织物的制作)在以tepa的浓度达到30mm、三乙基胺的浓度达到473mm的方式分别溶解于500ml的dmso中得到的液体中,浸渍10g的上述氯乙酰胺基甲基化针织物d,在40℃反应3小时。将该针织物用dmso洗涤3次后,浸渍于以对氯苯基异氰酸酯的浓度达到20mm的方式溶解于500ml的dmso中得到的液体,在30℃反应1小时。然后,由反应液取出针织物,浸渍于与反应液相同量的dmso中进行洗涤,接着用甲醇洗涤,接着浸渍于水中进行洗涤,得到四亚乙基五胺
‑
对氯苯基化针织物(以下称为吸附材料10)。
[0105]
(以十六烷基作为配体的颗粒的取出)将以十六烷基为配体的纤维素颗粒作为吸附体的吸附型血液净化器
“リクセル”
(注册商标;株式会社
カネカ
公司制)解体,取出填充于柱内部的颗粒(以下称为吸附材料11)。
[0106]
(二甲基辛基铵化无纺布的制作)在将n,n
‑
二甲基辛基胺50g和碘化钾8g溶解于360ml的n,n
‑
二甲基甲酰胺得到的溶液中,浸渍5g氯乙酰胺基甲基化无纺布e,在85℃的浴中加热3小时。将该无纺布用异丙醇
洗涤后,浸渍于1mol/l的食盐水中后,进行水洗,得到二甲基辛基铵化无纺布(以下称为吸附材料12)。
[0107]
(吸附材料1的峰半径的算出)将约6mg浸渗于水的吸附材料1在将要进行dsc测定前取出,除去过量的表面附着水后,将吸附材料1封入铝制密封型样品容器中。接着,使用dsc q100(ta instruments公司制),湿润状态下骤冷至
‑
55℃,以0.3℃/分钟升温至5℃,测定差示扫描热量,将峰顶温度作为熔点,得到dsc曲线。要说明的是,温度、热量校正使用纯水。由得到的dsc曲线,按照ishikiriyama等人的方法(journal of colloid and interface science,1995年,171卷,p.92
‑
102),制作微孔孔径分布曲线。微孔孔径分布曲线是横轴为微孔半径、竖轴为每1g湿润了的吸附材料的微孔体积进行作图得到的。在此,峰半径是上述微孔孔径分布曲线中,每1g冷冻干燥了的吸附材料的微孔体积取最大值的微孔半径。吸附材料1的峰半径示于表1。
[0108]
(吸附材料1的zeta电位的测定)将浸渗于水的吸附材料1在将要进行zeta电位的测定前取出,除去过量的表面附着水。zeta电位的测定使用固体表面分析用zeta电位测定装置surpass tm
(
アントンパール
公司制)测定。zeta电位的值采用在ph7.4、温度20
±
5℃条件下测定的值。吸附材料1的zeta电位示于表1。
[0109]
(吸附材料1的纤维直径的测定)将吸附材料1冷冻包埋,用切片机制作截面。对得到的观察面进行导电处理,作为观察样品。接着使用场发射型扫描电子显微镜s
‑
5500(日立
ハイテクノロジーズ
公司制)对观察样品的截面进行随机观察,拍摄截面图像100点。对得到的截面图像中的纤维截面算出直径。该操作对截面图像全部100个点进行,得到的各直径的平均值作为吸附材料1的纤维直径。结果示于表1。
[0110]
(吸附材料1的非多孔部分的面积的比例b的算出)将吸附材料1通过四氧化钌染色冷冻超薄切片法进行前处理,通过透射型电子显微镜em
‑
1400plus(日本电子公司制),在加速电压100kv的条件下以8000倍的倍率随机观察包括吸附材料1的中心的垂直于长度方向的截面,拍摄截面图像100点。在拍摄的截面图像中,以非多孔部分的面积为a1、多孔部分的面积为a2的情况下,求出通过式(1)求出的非多孔部分的面积的比例b。非多孔部分与多孔部分的辨别通过截面图像中存在的材料表面的微孔判断。非多孔部分的面积是指存在小于10nm的微孔的区域的面积和不存在微孔的区域的面积之和,多孔部分的面积是存在10nm以上微孔的区域的面积。面积的算出使用图像编辑软件(photoshop elements.14.0 adobe公司制)算出。吸附材料1的非多孔部分的面积的比例b示于表1。
[0111]
b=a1/(a1 a2)
×
100
ꢀ・・・
式(1)(吸附材料1的微孔体积的算出)将约6mg浸渗于水的吸附材料1在将要进行dsc测定前取出,除去过量的表面附着水后,将吸附材料1封入铝制密封型样品容器中。接着,使用dsc q100(ta instruments公司制),在湿润状态下骤冷至
‑
55℃,以0.3℃/分钟升温至5℃,测定差示扫描热量,将峰顶温度作为熔点,得到dsc曲线。要说明的是,温度、热量校正使用纯水。由得到的dsc曲线,按照ishikiriyama等人的方法(journal of colloid and interface science,1995年,171卷,
p.92
‑
102),算出吸附材料1的微孔体积。结果示于表1。
[0112]
(吸附材料2~12的峰半径的算出)通过进行与吸附材料1同样的操作,分别算出吸附材料2~12的峰半径。结果示于表1或表2。
[0113]
(吸附材料2~12的zeta电位的测定)通过进行与吸附材料1同样的操作,分别算出吸附材料2~12的zeta电位。结果示于表1或表2。
[0114]
(吸附材料2~10和吸附材料12的纤维直径的测定)通过进行与吸附材料1同样的操作,算出吸附材料2~10和吸附材料12的纤维直径。结果示于表1或表2。
[0115]
(吸附材料11的颗粒直径的测定)将吸附材料11冷冻包埋,通过切片机制作截面。对得到的观察面进行导电处理,作为观察样品。接着,使用场发射型扫描电子显微镜s
‑
5500(日立
ハイテクノロジーズ
公司制),对观察样品的截面进行随机观察,拍摄截面图像100点。对得到的截面图像中的纤维截面制作最小内包圆,算出最小内包圆的直径。该操作对截面图像所有100点进行,得到的各直径的平均值作为吸附材料11的颗粒直径。结果示于表2。
[0116]
(吸附材料2~12的非多孔部分的面积的比例b的算出)通过进行与吸附材料1同样的操作,算出吸附材料2~12的非多孔部分的面积的比例b。结果示于表1或表2。
[0117]
(吸附材料2~12的微孔体积的算出)通过进行与吸附材料1同样的操作,算出吸附材料2~12的微孔体积。结果示于表1或表2。
[0118]
(实施例1)1.可溶性肿瘤坏死因子受体的吸附能力的评价:在含有可溶性肿瘤坏死因子受体的液体中浸渍可溶性肿瘤坏死因子受体的吸附材料规定时间后取出,由浸渍前后的液体中的可溶性肿瘤坏死因子受体浓度之差测定可溶性肿瘤坏死因子受体的吸附率。以下示出测定方法。要说明的是,作为可能性肿瘤坏死因子受体使用可溶性肿瘤坏死因子受体1(stnfr1)。
[0119]
将吸附材料1切成直径6mm的圆板状后,将其每8片放入聚丙烯制的容器中。在该容器中,将以可溶性肿瘤坏死因子受体1(recombinant human tnfri/tnfrsf1a,636
‑
r1,r&d systems(注册商标),inc.)的浓度达到10000pg/ml方式制备的胎牛血清(fetal bovine serum,以下称为fbs)相对于0.055cm 3
的吸附材料1添加1ml,在37℃的培养器内翻转混合2小时后,通过酶联免疫吸附(elisa:quantikine(注册商标) elisa human tnf ri/tnfrsf1a immunoassay,drt100,r&d systems(注册商标),inc.)法测定fbs中的stnfr1浓度。由翻转混合前和翻转混合后的stnfr1浓度通过下式2算出stnfr1的吸附率。结果示于表1。
[0120]
吸附材料1的stnfr1的吸附率(%)={翻转混合前的stnfr1浓度(pg/ml)
‑
翻转混合后的stnfr1浓度(pg/ml)}/翻转混合前的stnfr1浓度(pg/ml)
×
100
ꢀ・・・
式22.血小板的吸附能力的评价:
在填充了吸附材料1的微型柱中通入健康人志愿者的血液,由通液前后的血液中的血小板数的差测定血小板吸附率。以下示出测定方法。
[0121]
在上下具有液体的出入口的圆筒状柱(内径1cm
×
高度1.2cm,内容积0.94cm3,外径2cm,聚碳酸酯制)中,层叠填充切成直径1cm的圆板状的吸附材料1,由此制作填充了吸附材料1的柱。将添加了lps达到70eu/ml的健康人志愿者血液(采血时每1ml血液添加0.13mg甲磺酸萘莫司他(
メチル
酸
ナファモスタット
))在37℃、30分钟、65rpm的条件下在热水浴中振荡,将血液用泵以0.63ml/min的流量通入该柱中,在柱入口和出口分别采取血液的样品。作为柱入口的样品,采取浸渍于热水浴中的血液(柱通液前的血液)。作为柱出口的样品,将血液流入柱内的时间点作为0分钟时,采取经过3.5分钟至6.5分钟后的血液(柱通液后的血液)。使用多项目自动血球分析装置xt
‑
1800i(sysmex公司制),分别测定采取的血液中含有的血小板数。接着,通过下式3算出血小板吸附率。结果示于表1。
[0122]
血小板吸附率(%)=(柱通液前的血小板数
‑
柱通液后的血小板数)/柱通液前的血小板数
×
100
ꢀ・・・
式3(实施例2)使用吸附材料2,通过进行与实施例1同样的测定,测定stnfr1的吸附率、血小板吸附率。结果示于表1。
[0123]
(实施例3)使用吸附材料3,通过进行与实施例1同样的测定,测定stnfr1的吸附率、血小板吸附率。结果示于表1。
[0124]
(实施例4)使用吸附材料4,通过进行与实施例1同样的测定,测定stnfr1的吸附率、血小板吸附率。结果示于表1。
[0125]
(实施例5)使用吸附材料5,通过进行与实施例1同样的测定,测定stnfr1的吸附率、血小板吸附率。结果示于表1。
[0126]
(实施例6)使用吸附材料6,通过进行与实施例1同样的测定,测定stnfr1的吸附率、血小板吸附率。结果示于表1。
[0127]
(比较例1)使用吸附材料7,通过进行与实施例1同样的测定,测定stnfr1的吸附率、血小板吸附率。结果示于表2。
[0128]
(比较例2)使用吸附材料8,通过进行与实施例1同样的测定,测定stnfr1的吸附率、血小板吸附率。结果示于表2。
[0129]
(比较例3)使用吸附材料9,通过进行与实施例1同样的测定,测定stnfr1的吸附率、血小板吸附率。结果示于表2。
[0130]
(比较例4)使用吸附材料10,通过进行与实施例1同样的测定,测定stnfr1的吸附率、血小板
吸附率。结果示于表2。
[0131]
(比较例5)使用吸附材料11,通过进行与实施例1同样的测定,测定stnfr1的吸附率、血小板吸附率。结果示于表2。
[0132]
(比较例6)使用吸附材料12,通过进行与实施例1同样的测定,测定stnfr1的吸附率、血小板吸附率。结果示于表2。
[0133]
[表1]
[0134]
表1中,“峰半径”表示由通过差示扫描量热仪测定的熔点分布算出的峰半径,“zeta电位”表示ph7.4下的zeta电位,“非多孔部分的面积的比例b”表示在包含不溶性高分子材料的中心的垂直于长度方向的截面中,以非多孔部分的面积为a1,以多孔部分的面积为a2的情况下,由式(1)求出的非多孔部分的面积的比例,“微孔体积”表示由通过差示扫描量热仪测定的熔点分布算出的微孔体积,“stnfr1吸附率”表示可溶性肿瘤坏死因子受体1(stnfr1)的吸附率。
[0135]
[表2]
[0136]
表2中的各项目表示与表1同样的项目。
[0137]
由表1和表2的结果可知,本发明的吸附材料的可溶性肿瘤坏死因子受体的吸附性能优异。另外,可知也可适当抑制对生物体的止血功能有用的血小板的吸附。
[0138]
产业实用性本发明的吸附材料可高效率地吸附可溶性肿瘤坏死因子受体,因此可用于医疗领域中的生物体成分处理、特别是血液成分处理用途。
[0139]
符号说明1:微孔体积的最大值2:峰半径3:纤维形状的水不溶性高分子材料4:中心5:表示垂直于长度方向的符号6:包括中心的垂直于长度方向的截面7:垂直于长度方向的方向的截面上的中心线8:圆球颗粒形状的水不溶性高分子材料9:椭圆体颗粒形状的水不溶性高分子材料10:膜形状的水不溶性高分子材料11:多孔部分
12:非多孔部分
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。