1.本发明涉及树脂发泡体及发泡构件。
背景技术:
2.出于保护移动设备的电池、基板等构件的目的而使用了发泡体,但近年来,由于高容量的数据通信、应用的组合使用等,存在处理速度增加、各构件容易发热的倾向。因此,要求上述发泡体耐受在高温下长时间使用。
3.作为形成耐热性优异的发泡体的方法,考虑了使用高熔点(例如150℃以上)树脂而构成发泡体的方法。然而,在添加化学发泡剂(例如热分解型发泡剂)来赋予发泡性的情况下,有时在高熔点树脂的成型温度下发生发泡,难以使用高熔点树脂得到发泡体。
4.另一方面,对于使用发泡体的部分的间隙的大小,近年来要求对应更小的间隙。另外,在将发泡体应用于移动设备的情况下,由于设备的落下、来自外部的压力负荷,容易对各构件施加无法预测的负载。因此,如果能够使这样的负载的应力有效地分散,则能够吸收冲击,防止由无法预测的负载导致的电子设备的破坏。因此,要求能够对应更小的间隙、并且具有更高水平的应力分散性的发泡体。
5.作为不使用化学发泡剂而得到发泡体的方法,对于使不活泼气体在高压下溶解于聚合物后使压力急剧地降低而形成发泡结构的方法进行了研究。例如,在专利文献1中,公开了在压力容器中添加热塑性聚合物,一边加热至聚合物的软化点一边加入高压气体,然后使压力降低而形成气泡的方法。然而,专利文献1的发泡体虽然具有某种程度的柔软性,但不具有耐热性。另外,专利文献1中没有关于发泡体的应力分散性(冲击吸收性)的任何公开或启示。
6.另外,在专利文献2中公开了通过特定熔点的聚烯烃树脂、热塑性弹性体的选择而对聚烯烃类发泡体赋予耐热性的方法。然而,专利文献2中没有关于发泡体的应力分散性(冲击吸收性)的任何公开或启示。
7.现有技术文献
8.专利文献
9.专利文献1:日本特开平6
‑
322168号公报
10.专利文献2:日本特开2013
‑
082881公报
技术实现要素:
11.发明所解决的问题
12.本发明的课题在于,提供具有高应力分散性、并且耐热性优异的树脂发泡体。
13.解决问题的方法
14.本发明的树脂发泡体具有气泡结构,其表观密度为0.05g/cm3~0.50g/cm3、50%压缩载荷为2.0n/cm2~30n/cm2,并且所述树脂发泡体的表观密度d(g/cm3)和在650℃下的残渣r(%)满足下述式(1)的关系,
15.1≤{(100-r)/d}/100≤10
···
(1)。
16.在一个实施方式中,上述气泡的平均气泡直径为10μm~200μm。
17.在一个实施方式中,上述气泡的气泡直径的变动系数为0.5以下。
18.在一个实施方式中,上述气泡结构中的气泡率为30%以上。
19.在一个实施方式中,上述气泡结构中的气泡壁的厚度为0.1μm~10μm。
20.在一个实施方式中,上述树脂发泡体在23℃下的拉伸模量为0.6mpa以上。
21.在一个实施方式中,上述树脂发泡体的应力保持力为60%以上。
22.在一个实施方式中,上述树脂发泡体包含填充材料。
23.在一个实施方式中,上述填充材料为无机物。
24.在一个实施方式中,上述填充材料为有机物。
25.在一个实施方式中,构成上述树脂发泡体的树脂为聚烯烃类树脂。
26.在一个实施方式中,上述聚烯烃类树脂是聚烯烃类弹性体以外的聚丙烯与聚烯烃类弹性体的混合物。
27.在一个实施方式中,在上述树脂发泡体的一面或两面具有热熔融层。
28.根据本发明的另一方面,提供一种发泡构件,该发泡构件具备:由上述树脂发泡体构成的树脂发泡层、和配置于该树脂发泡层的至少一侧的粘合剂层。
29.发明的效果
30.根据本发明,能够提供具有高应力分散性、并且耐热性优异的树脂发泡体。
附图说明
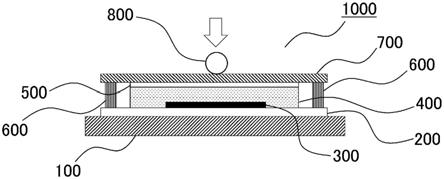
31.图1是应力缓和试验机的剖面示意图。
32.符号说明
33.应力缓和试验机
ꢀꢀꢀꢀꢀ
1000
34.铁制的支撑体
ꢀꢀꢀꢀꢀꢀꢀ
100
35.聚碳酸酯板
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
200
36.应力测定膜
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
300
37.树脂发泡结构体
ꢀꢀꢀꢀꢀ
400
38.双面粘接带
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
500
39.间隔件
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
600
40.abs板
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
700
41.铁球
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
800
具体实施方式
42.《《1.树脂发泡体》》
43.本发明的树脂发泡体具有气泡结构,其表观密度为0.05g/cm3~0.50g/cm3,50%压缩载荷为2.0n/cm2~30n/cm2,并且该树脂发泡体的表观密度d(g/cm3)和在650℃下的残渣r(%)满足下述式(1)的关系。
44.1≤{(100-r)/d}/100≤10
···
(1)
45.需要说明的是,在本说明书中,残渣r是指在氮气气氛中以升温速度20℃/min的条
件在25℃~680℃的测定范围内将树脂发泡体升温时在650℃下的残渣。残渣r例如能够使用sii nanotechnology公司制造的商品名“tg/dta6200”来测定。
46.本发明的树脂发泡体通过为上述构成而具有高应力分散性和耐热性。另外,本发明的树脂发泡体的柔软性也优异。本发明的树脂发泡体具有高应力分散性的一个原因在于,即使在间隙窄的位置也能够发挥优异的冲击吸收性。另外,耐热性优异的上述树脂发泡体能够适宜地用作高性能的移动设备等容易变成高温的设备中的保护构件。
47.本发明的树脂发泡体的表观密度优选为0.06g/cm3~0.45g/cm3、更优选为0.07g/cm3~0.40g/cm3、进一步优选为0.08g/cm3~0.35g/cm3。如果是这样的范围,则能够得到应力分散性更优异的树脂发泡体。表观密度的测定方法在后面叙述。
48.本发明的树脂发泡体的50%压缩载荷优选为2.5n/cm2~25n/cm2、更优选为3.0n/cm2~20n/cm2、进一步优选为3.5n/cm2~15n/cm2。如果是这样的范围,则能够得到应力分散性更优异的树脂发泡体。表观密度的测定方法在后面叙述。
49.如上所述,表观密度d(g/cm3)和在650℃下的残渣r(%)满足下述式(1)的关系。
50.1≤{(100-r)/d}/100≤10
···
(1)
51.优选表观密度d(g/cm3)和在650℃下的残渣r(%)满足下述式(2)的关系、更优选表观密度d(g/cm3)和在650℃下的残渣r(%)满足下述式(3)的关系、进一步优选表观密度d(g/cm3)和在650℃下的残渣r(%)满足下述式(4)的关系。如果表观密度d和残渣r为这样的关系,则能够得到高度地兼顾了应力分散性和耐热性的树脂发泡体。
52.2≤{(100-r)/d}/100≤9.5
···
(2)
53.3≤{(100-r)/d}/100≤8.5
···
(3)
54.3.5≤{(100-r)/d}/100≤8
···
(4)
55.本发明的树脂发泡体在650℃下的残渣r优选为10重量%以上、更优选为15重量%以上、进一步优选为20重量%以上、特别优选为25重量%、最优选为35重量%以上。如果是这样的范围,则能够得到耐热性特别优异的树脂发泡体。上述残渣r的上限例如为80重量%,在一个实施方式中为60重量%。在一个实施方式中,残渣r可以是树脂发泡体中所含的无机成分(例如无机填充材料)。
56.本发明的树脂发泡体具有气泡结构(气孔结构)。作为这样的气泡结构(气孔结构),可列举独立气泡结构、连续气泡结构、半连续半独立气泡结构(混合存在有独立气泡结构和连续气泡结构的气泡结构)等。本发明的树脂发泡体的气泡结构优选为连续气泡结构、半连续半独立气泡结构,更优选为半连续半独立气泡结构。在本发明的树脂发泡体的气泡结构为半连续半独立气泡结构的情况下,其中的独立气泡结构的比例优选为40%以下、更优选为30%以下。
57.本发明的树脂发泡体的独立气泡率例如如下所述地求出:在温度23℃、湿度50%的环境中,使测定对象在水分中下沉,测定之后的质量,然后在80℃的烘箱中使其充分地干燥后,再次测定质量。另外,如果是连续气泡,则能够保持水分,因此,可作为连续气泡测定其质量而求出。
58.上述气泡的平均气泡直径(平均气孔直径)优选为10μm~200μm、更优选为15μm~180μm、进一步优选为20μm~150μm、特别优选为23μm~120μm、尤其优选为25μm~100μm。如果是这样的范围,则能够得到柔软性及应力分散性更优异的树脂发泡体。另外,能够得到压
缩回复性也优异、并且对于重复冲击的耐性优异的树脂发泡体。平均气泡直径的测定方法在后面叙述。
59.上述气泡的气泡直径(气孔直径)的变动系数优选为0.5以下、更优选为0.48以下、进一步优选为0.45以下、特别优选为0.43以下,最优选小于0.4。如果是这样的范围,则由冲击导致的变形变得均匀,可防止局部的应力负荷,能够得到应力分散性优异、并且耐冲击性特别优异的树脂发泡体。该变动系数越小越优选,其下限例如为0.2(优选为0.15、更优选为0.1、进一步优选为0.01)。气泡直径的变动系数的测定方法在后面叙述。
60.上述气泡结构的气泡率(气孔率)优选为30%以上、更优选为50%以上、进一步优选为80%以上。如果是这样的范围,则能够得到压缩时的排斥应力小的树脂发泡体。对于这样的树脂发泡体而言,在间隙窄的位置稍微进行压缩并应用上述树脂发泡体时,能够减小对其它构件施加的应力。例如,在将树脂发泡体用于显示构件的情况下,能够使对该显示构件施加的应力松弛/分散,因此,从颜色不均减少、构件保护的观点考虑是有用的。该气泡率的上限例如为99%以下。气泡率的测定方法在后面叙述。
61.上述气泡结构中的气泡壁(气孔壁)的厚度优选为0.1μm~10μm、更优选为0.3μm~8μm、进一步优选为0.5μm~5μm、特别优选为0.7μm~4μm,最优选为1μm~3μm。如果是这样的范围,则能够得到柔软性及应力分散性更优异的树脂发泡体。气泡壁的厚度过薄时,树脂发泡体容易对于负载而变形,存在无法得到充分的应力分散效果的担忧。气泡壁的厚度过厚时,树脂发泡体变得难以对于负载而变形,在设备的间隙中使用时,存在高度差追随性变差的担忧。气泡壁的厚度的测定方法如下所述:通过导入树脂发泡体的气泡部的放大图像,使用相同计测器的解析软件,进行图像解析而进行测定。
62.本发明的树脂发泡体在23℃下的断裂伸长率优选为120%以下、更优选为110%以下、进一步优选为105%以下、更进一步优选为100%以下、特别优选为95%以下、最优选为90%以下。如果是这样的范围,则能够得到应力分散性优异、薄型且冲击吸收性优异的树脂发泡体。在拉伸试验中的断裂伸长率小时,对树脂发泡体施加负载时,该树脂发泡体的气孔壁的变形变小,例如在添加了填料的情况下,在构成该树脂发泡体的树脂与该填料的界面容易发生滑动,能进一步缓和负载。上述断裂伸长率的下限优选为1%以上、更优选为5%以上、进一步优选为10%以上、特别优选为15%以上、最优选为20%以上。另一方面,在拉伸试验中的断裂伸长率过大时,树脂发泡体的气孔壁的变形变大,存在变得难以缓和负载的担忧。断裂伸长率可以基于jis k 6767来测定。
63.将上述树脂发泡体放置于120℃的环境中500小时时的尺寸变化率优选为1%以下、更优选为0.8%以下。尺寸变化率越小越优选,其下限在现实中为0.1%(优选为0.05%)。上述尺寸变化率的测定方法在后面叙述。
64.上述树脂发泡体在23℃下的拉伸模量优选为0.6mpa以上、更优选为0.7mpa~5mpa、进一步优选为1mpa~4mpa。如果是这样的范围,则能够得到应力分散性优异、即使为薄膜也能够发挥优异的冲击吸收性的树脂发泡体。上述拉伸模量的测定方法在后面叙述。
65.上述树脂发泡体的应力保持力优选为60%以上、更优选为63%~100%、进一步优选为63%~95%。如果是这样的范围,则能够得到应力分散性优异、即使为薄膜也能够发挥优异的冲击吸收性的树脂发泡体。在本说明书中,上述应力保持力是指,以300m/min的速度将树脂发泡体(宽10mm
×
长100mm)沿着长度方向拉伸20%、刚拉伸后的拉伸强度与拉伸后
保持了120秒钟后的拉伸强度的比率(保持120秒钟后的拉伸强度/拉伸直后的应力保持力
×
100)。
66.作为本发明的树脂发泡体的形状,可以根据目的采用任意适当的形状。作为这样的形状,代表性地为片状,在该情况下,能够将本发明的树脂发泡体作为树脂发泡层处理。
67.在本发明的树脂发泡体的形状为片状的情况(即,树脂发泡层的情况)下,其厚度优选为30μm~5000μm、更优选为35μm~4000μm、进一步优选为40μm~3000μm、特别优选为45μm~2500μm。本发明的树脂发泡体即使为薄型,也能够发挥优异的冲击吸收性。这样的树脂发泡体能够适宜地用作应用于微小间隙的保护材料。
68.可以在本发明的树脂发泡体的一面或两面具有热熔融层。具有热熔融层的树脂发泡体例如能够通过使用加热至构成树脂发泡体的树脂组合物的熔融温度以上的一对加热辊对树脂发泡体(或树脂发泡体的前体)进行压延而得到。
69.在不损害本发明效果的范围内,本发明的树脂发泡体可以通过任意适当的方法形成。作为这样的方法,代表性地,可举出使包含树脂材料(聚合物)的树脂组合物发泡的方法。
70.《1
‑
1.树脂组合物》
71.本发明的树脂发泡体包含任意适当的树脂。上述树脂发泡体代表性地可以通过使包含树脂的组合物(树脂组合物)发泡而得到。
72.作为构成上述树脂发泡体树脂(即,上述树脂组合物中所含的树脂),可以在不损害本发明效果的范围内使用任意适当的树脂。作为该树脂,可列举例如:丙烯酸类树脂、有机硅类树脂、氨基甲酸酯类树脂、聚烯烃类树脂、酯类树脂、橡胶类树脂等。上述树脂可以仅为一种,也可以为两种以上。
73.相对于树脂发泡体100重量份,上述树脂的含有比例优选为30重量份~95重量份、更优选为35重量份~90重量份、进一步优选为40重量份~80重量份、特别优选为40重量份~60重量份。
74.在一个实施方式中,上述树脂发泡体包含聚烯烃类树脂。聚烯烃类树脂可以仅为一种,也可以为两种以上。
75.相对于树脂发泡体100重量份,聚烯烃类树脂的含有比例优选为50重量份~100重量份、更优选为70重量份~100重量份、进一步优选为90重量份~100重量份、特别优选为95重量份~100重量份。
76.作为聚烯烃类树脂,可优选列举选自聚烯烃及聚烯烃类弹性体中的至少一种,更优选为将聚烯烃与聚烯烃类弹性体组合使用的方式。聚烯烃可以仅为一种,也可以为两种以上。聚烯烃类弹性体可以仅为一种,也可以为两种以上。需要说明的是,在本说明书中,在称为“聚烯烃”的情况下,不包括“聚烯烃类弹性体”。
77.在将聚烯烃与聚烯烃类弹性体作为聚烯烃类树脂组合使用的情况下,聚烯烃与聚烯烃类弹性体的含有比率(聚烯烃/聚烯烃类弹性体)以重量比率计优选为1/99~99/1、更优选为10/90~90/10、进一步优选为20/80~80/20、特别优选为30/70~70/30。
78.作为聚烯烃,可以在不损害本发明效果的范围内采用任意适当的聚烯烃。作为这样的聚烯烃,可列举例如:直链状的聚烯烃、支链状的(具有支链的)聚烯烃等。
79.作为这样的聚烯烃,可列举例如:包含α
‑
烯烃而构成的聚合物、即在1分子中至少
具有来自α
‑
烯烃的结构单元的聚合物。这样的聚烯烃可以为仅由α
‑
烯烃构成的聚合物,也可以为由α
‑
烯烃和α
‑
烯烃以外的单体成分构成的聚合物。
80.聚烯烃可以为均聚物,也可以为包含两种以上单体的共聚物。在聚烯烃为共聚物的情况下,作为其共聚方式,可采用任意适当的共聚方式。作为这样的共聚方式,可列举例如:无规共聚物、嵌段共聚物等。
81.作为能够构成聚烯烃的α
‑
烯烃,优选例如碳原子数2~8的α
‑
烯烃(例如乙烯、丙烯、1
‑
丁烯、1
‑
戊烯、1
‑
己烯、4
‑
甲基
‑1‑
戊烯、1
‑
庚烯、1
‑
辛烯等)。能够构成聚烯烃的α
‑
烯烃可以仅为一种,也可以为两种以上。
82.作为能够构成聚烯烃的α
‑
烯烃以外的单体成分,可列举例如:乙酸乙烯酯、丙烯酸、丙烯酸酯、甲基丙烯酸、甲基丙烯酸酯、乙烯醇等烯属不饱和单体。能够构成聚烯烃的α
‑
烯烃以外的单体成分可以仅为一种,也可以为两种以上。
83.作为聚烯烃,具体可列举例如:低密度聚乙烯、中密度聚乙烯、高密度聚乙烯、直链状低密度聚乙烯、聚丙烯(丙烯均聚物)、乙烯与丙烯的共聚物、乙烯与乙烯以外的α
‑
烯烃的共聚物、丙烯与丙烯以外的α
‑
烯烃的共聚物、乙烯、丙烯及除乙烯和丙烯以外的α
‑
烯烃的共聚物、丙烯与烯属不饱和单体的共聚物等。
84.作为聚烯烃,从能够进一步展现本发明效果的方面考虑,优选为以丙烯为必须的单体成分而构成的聚合物(聚丙烯类聚合物)、即至少具有来自丙烯的结构单元的聚合物。作为这样的聚丙烯类聚合物,可列举例如:聚丙烯(丙烯均聚物)、乙烯与丙烯的共聚物、丙烯与丙烯以外的α
‑
烯烃的共聚物等,优选为聚丙烯(丙烯均聚物)。聚丙烯类聚合物可以仅为一种,也可以为两种以上。
85.从能够进一步展现本发明效果的方面考虑,聚烯烃在温度230℃下的熔体流动速率(mfr)优选为0.2g/10分~10g/10分、更优选为0.25g/10分~5g/10分、进一步优选为0.3g/10分~3g/10分、特别优选为0.35g/10分~1.5g/10分。需要说明的是,聚烯烃在温度230℃下的熔体流动速率(mfr)是指基于iso1133(jis
‑
k
‑
7210)在温度230℃、负载2.16kgf的条件下测定的mfr。
86.作为聚烯烃,从能够进一步展现本发明效果的方面考虑,优选组合使用在温度230℃下的熔体流动速率(mfr)在上述的范围内的两种以上不同的聚烯烃。在该情况下,组合使用在温度230℃下的熔体流动速率(mfr)优选为0.2g/10分以上且小于0.7g/10分(更优选为0.2g/10分~0.65g/10分)的聚烯烃、与在温度230℃下的熔体流动速率(mfr)优选为0.7g/10分~10g/10分(更优选为0.7g/10分~5g/10分、进一步优选为0.7g/10分~3g/10分、特别优选为0.7g/10分~1.5g/10分、最优选为0.7g/10分~1.3g/10分)的聚烯烃。
87.作为聚烯烃,在组合使用在温度230℃下的熔体流动速率(mfr)为上述的范围内的两种以上不同的聚烯烃的情况下,从能够进一步展现本发明效果的方面考虑,例如上述在温度230℃下的熔体流动速率(mfr)优选为0.2g/10分以上且小于0.7g/10分(更优选为0.2g/10分~0.65g/10分)的聚烯烃、与在温度230℃下的熔体流动速率(mfr)优选为0.7g/10分~10g/10分(更优选为0.7g/10分~5g/10分、进一步优选为0.7g/10分~3g/10分、特别优选为0.7g/10分~1.5g/10分、最优选为0.7g/10分~1.3g/10分)的聚烯烃的含有比率以重量比率计优选为1/99~99/1、更优选为10/90~90/10、进一步优选为20/80~80/20、特别优选为30/70~70/30、最优选为40/60~60/40。
88.作为聚烯烃,可以使用市售品,可列举例如:“e110g”(普瑞曼聚合物株式会社制)、“ea9”(日本聚丙烯株式会社制)、“ea9ft”(日本聚丙烯株式会社制)、“e
‑
185g”(普瑞曼聚合物株式会社制)、“wb140hms”(borealis公司制)、“wb135hms”(borealis公司制)等。
89.作为聚烯烃类弹性体,可以在不损害本发明效果的范围内采用任意适当的聚烯烃类弹性体。作为这样的聚烯烃类弹性体,可列举例如:乙烯
‑
丙烯共聚物、乙烯
‑
丙烯
‑
二烯共聚物、乙烯
‑
乙酸乙烯酯共聚物、聚丁烯、聚异丁烯、氯化聚乙烯、使聚烯烃成分与橡胶成分物理分散而得到的弹性体、具有聚烯烃成分与橡胶成分发生了微相分离的结构的弹性体等所谓的非交联型的热塑性烯烃类弹性体(tpo);通过在存在交联剂的条件下对包含形成基体的树脂成分a(烯烃类树脂成分a)及形成域(domain)的橡胶成分b的混合物进行动态热处理而得到的动态交联型热塑性烯烃类弹性体(tpv);等等,该动态交联型热塑性烯烃类弹性体是具有在作为基体(海相)的树脂成分a中细密地分散有交联橡胶粒子作为域(岛相)的海岛结构的多相体系的聚合物。
90.聚烯烃类弹性体优选包含橡胶成分。作为这样的橡胶成分,可列举日本特开平08
‑
302111号公报、日本特开2010
‑
241934号公报、日本特开2008
‑
024882号公报、日本特开2000
‑
007858号公报、日本特开2006
‑
052277号公报、日本特开2012
‑
072306号公报、日本特开2012
‑
057068号公报、日本特开2010
‑
241897号公报、日本特开2009
‑
067969号公报、日本再表03/002654号公报等中记载的橡胶成分。
91.作为具有聚烯烃成分与烯烃类橡胶成分发生了微相分离的结构的弹性体,具体可列举:由聚丙烯树脂(pp)和乙丙橡胶(epm)形成的弹性体、由聚丙烯树脂(pp)和乙烯
‑
丙烯
‑
二烯橡胶(epdm)形成的弹性体等。从相容性的观点考虑,聚烯烃成分与烯烃类橡胶成分的重量比以聚烯烃成分/烯烃类橡胶计优选为90/10~10/90、更优选为80/20~20/80。
92.对于动态交联型热塑性烯烃类弹性体(tpv)而言,一般来说,与非交联型的热塑性烯烃类弹性体(tpo)相比,其弹性模量高,并且压缩永久应变也小。由此,回复性良好,在制成发泡体的情况下,能够显示出优异的回复性。
93.动态交联型热塑性烯烃类弹性体(tpv)是指,如上述那样通过在存在交联剂的条件下对包含形成基体的树脂成分a(烯烃类树脂成分a)及形成域的橡胶成分b的混合物进行动态热处理而得到的具有在作为基体(海相)的树脂成分a中细密地分散有交联橡胶粒子作为域(岛相)的海岛结构的多相体系的聚合物。
94.作为动态交联型热塑性烯烃类弹性体(tpv),可列举例如:日本特开2000
‑
007858号公报、日本特开2006
‑
052277号公报、日本特开2012
‑
072306号公报、日本特开2012
‑
057068号公报、日本特开2010
‑
241897号公报、日本特开2009
‑
067969号公报、日本再表03/002654号等中记载的动态交联型热塑性烯烃类弹性体。
95.作为动态交联型热塑性烯烃类弹性体(tpv),可使用市售品,可列举例如:“zeotherm”(日本瑞翁株式会社制)、“thermorun”(三菱化学株式会社制)、“sarlink 3245d”(东洋纺织株式会社制)等。
96.聚烯烃类弹性体在温度230℃下的熔体流动速率(mfr)优选为2g/10分~15g/10分、更优选为3g/10分~10g/10分、进一步优选为3.5g/10分~9g/10分、特别优选为4g/10分~8g/10分、最优选为4.5g/10分~7.5g/10分。需要说明的是,聚烯烃类弹性体在温度230℃下的熔体流动速率(mfr)是指基于iso1133(jis
‑
k
‑
7210)在温度230℃、负载2.16kgf的条件
下测定的mfr。
97.聚烯烃类弹性体的熔融张力(190℃、断裂时)优选小于10cn、更优选为5cn~9.5cn。
98.聚烯烃类弹性体的jis a硬度优选为30
°
~95
°
、更优选为35
°
~90
°
、进一步优选为40
°
~88
°
、特别优选为45
°
~85
°
、最优选为50
°
~83
°
。需要说明的是,jis a硬度是指基于so7619(jis k6253)测定的硬度。
99.在一个实施方式中,上述树脂发泡体(即树脂组合物)可以进一步包含填充材料。通过含有填充材料,能够形成需要大的能量使气泡壁(气孔壁)变形的树脂发泡体,该树脂发泡体发挥优异的冲击吸收性。另外,通过含有填充材料,能够形成微细且均匀的气泡结构,在能够展现优异的冲击吸收性这方面也是有利的。填充材料可以单独使用仅一种,也可以组合使用两种以上。
100.相对于构成树脂发泡体的聚合物100重量份,上述填充材料的含有比例优选为10重量份~150重量份、更优选为30重量份~130重量份、进一步优选为50重量份~100重量份。如果是这样的范围,则上述效果变得显著。
101.在一个实施方式中,上述填充材料为无机物。作为构成作为无机物的填充材料的材料,可列举例如:氢氧化铝、氢氧化镁、碳酸钙、碳酸镁、硅酸钙、硅酸镁、氧化钙、氧化镁、氧化铝、氮化铝、硼酸铝晶须、氮化硅、氮化硼、结晶二氧化硅、非晶质二氧化硅、金属(例如,金、银、铜、铝、镍)、碳、石墨等。
102.在一个实施方式中,上述填充材料为有机物。作为构成作为有机物的填充材料的材料,可列举例如:聚甲基丙烯酸甲酯(pmma)、聚酰亚胺、聚酰胺酰亚胺、聚醚醚酮、聚醚酰亚胺、聚酯酰亚胺等。
103.作为上述填充材料,可使用阻燃剂。作为阻燃剂,可列举例如:溴类阻燃剂、氯类阻燃剂、磷类阻燃剂、锑类阻燃剂等。从安全性的观点考虑,优选使用无卤无锑类阻燃剂。
104.作为无卤无锑类阻燃剂,可列举例如:包含铝、镁、钙、镍、钴、锡、锌、铜、铁、钛、硼等的化合物。作为这样的化合物(无机化合物),可列举例如:氢氧化铝、氢氧化镁、氧化镁/氧化镍的水合物、氧化镁/氧化锌的水合物等水合金属化合物等。
105.可以对上述填充材料实施任意适当的表面处理。作为这样的表面处理,可列举例如:硅烷偶联处理、硬脂酸处理等。
106.上述填充材料的堆积密度优选为0.8g/cm3以下、更优选为0.6g/cm3以下、进一步优选为0.4g/cm3以下、特别优选为0.3g/cm3以下。如果是这样的范围,则能够以良好的分散性含有填充材料,即使减少填充材料的含量,也能够充分地发挥填充材料添加效果。填充材料的含量少的树脂发泡体在高发泡、柔软、且应力分散性及外观优异的方面是有利的。填充材料的堆积密度的下限值例如为0.01g/cm3、优选为0.05g/cm3、更优选为0.1g/cm3。
107.上述填充材料的数均粒径(初级粒径)优选为5μm以下、更优选为3μm以下、进一步优选为1μm以下。如果是这样的范围,则能够以良好的分散性含有填充材料,并且能够形成均匀的气泡结构。其结果是,能够得到应力分散性及外观优异的树脂发泡体。填充材料的数均粒径的下限值例如为0.1μm。填充材料的数均粒径可以将在水100g中混合填充剂1g而制备的悬浮液作为样品、并使用粒度分布计(micrtracii、microtrac bel株式会社)来测定。
108.上述填充材料的比表面积优选为2m2/g以上、更优选为4m2/g以上、进一步优选为
6m2/g以上。如果是这样的范围,则能够以良好的分散性含有填充材料,并且能够形成均匀的气泡结构。其结果是,能够得到应力分散性及外观优异的树脂发泡体。填充材料的比表面积的上限值例如为20m2/g。填充材料的比表面积能够通过利用bet法来测定,即通过使吸附占有面积已知的分子在利用液氮的低温下吸附于填充材料表面,根据其吸附量来测定。
109.可以在不损害本发明效果的范围内在树脂组合物中包含任意适当的其它成分。这样的其它成分可以仅为一种,也可以为两种以上。作为这样的其它成分,可列举例如:橡胶、作为树脂材料配合的聚合物以外的树脂、软化剂、脂肪族类化合物、防老化剂、抗氧剂、光稳定剂、耐候剂、紫外线吸收剂、分散剂、增塑剂、碳、抗静电剂、表面活性剂、交联剂、增稠剂、防锈剂、有机硅类化合物、张力改性剂、防收缩剂、流动性改性剂、凝胶化剂、固化剂、加强剂、发泡剂、发泡成核剂、着色剂(颜料、染料等)、ph调节剂、溶剂(有机溶剂)、热聚合引发剂、光聚合引发剂、润滑剂、结晶成核剂、结晶化促进剂、硫化剂、表面处理剂、分散助剂等。
110.《1
‑
2.树脂发泡体的形成》
111.本发明的树脂发泡体代表性地通过使树脂组合物发泡而得到。作为发泡的方法(气泡的形成方法),可采用物理方法、化学方法等发泡成型中通常利用的方法。即,本发明的树脂发泡体代表性地可以是通过物理方法进行发泡而形成的发泡体(物理发泡体),也可以是通过化学方法进行发泡而形成的发泡体(化学发泡体)。物理方法一般是通过使空气、氮等气体成分分散于聚合物溶液并通过机械混合形成气泡的方法(机械发泡体)。化学方法一般是通过利用添加于聚合物基体的发泡剂的热分解产生的气体形成气孔而得到发泡体的方法。
112.上述树脂组合物例如可以通过用任意适当的熔融混炼装置、例如开放型的混合辊、非开放型的班伯里混合机、单螺杆挤出机、双螺杆挤出机、连续式混炼机、加压捏合机等任意适当的机构将构成成分混合而制备。
113.<形成本发明的树脂发泡体的实施方式1>
114.作为形成本发明的树脂发泡体的一个实施方式1,例如可列举以下方式:经过使乳液树脂组合物(包含树脂材料等的乳液)机械发泡而起泡化的工序(工序a)来形成树脂发泡体。作为起泡装置,可列举例如:高速剪切方式的装置、振动方式的装置、加压气体的喷出方式的装置等。这些起泡装置中,从气泡直径的微细化、大容量制作的观点考虑,优选高速剪切方式的装置。形成本发明的树脂发泡体的该一个实施方式1无论是在由哪种树脂组合物形成时均可以应用。
115.从成膜性的观点考虑,优选乳液的固体成分浓度高的情况。乳液的固体成分浓度优选为30重量%以上、更优选为40重量%以上、进一步优选为50重量%以上。
116.通过机械搅拌进行起泡时的气泡是气体进入到乳液中而成的。作为气体,只要对于乳液无活性,就可以在不损害本发明效果的范围内采用任意适当的气体。作为这样的气体,可列举例如:空气、氮气、二氧化碳等。
117.经过将通过上述方法起泡化后的乳液树脂组合物(含气泡乳液树脂组合物)涂敷于基材上并进行干燥的工序(工序b),能够得到本发明的树脂发泡体。作为基材,可列举例如:进行了剥离处理的塑料膜(进行了剥离处理的聚对苯二甲酸乙二醇酯膜等)、塑料膜(聚对苯二甲酸乙二醇酯膜等)等。
118.在工序b中,作为涂敷方法、干燥方法,可以在在不损害本发明效果的范围内采用
任意适当的方法。工序b优选包括:将涂布于基材上的含气泡乳液树脂组合物以50℃以上且小于125℃的温度干燥的预干燥工序b1;和之后进一步以125℃以上且200℃以下的温度进行干燥的正式干燥工序b2。
119.通过设置预干燥工序b1和正式干燥工序b2,能够防止由急剧的温度上升导致的气泡的合为一体化、气泡的破裂。特别是对于厚度小的发泡片而言,由于温度的急剧的上升,气泡会合为一体化、破裂,因此,设置预干燥工序b1的意义很大。预干燥工序b1中的温度优选为50℃~100℃。预干燥工序b1的时间优选为0.5分钟~30分钟、更优选为1分钟~15分钟。正式干燥工序b2中的温度优选为130℃~180℃、更优选为130℃~160℃。正式干燥工序b2的时间优选为0.5分钟~30分钟、更优选为1分钟~15分钟。
120.<形成本发明的树脂发泡体的实施方式2>
121.作为形成本发明的树脂发泡体的一个实施方式2,可举出利用发泡剂使树脂组合物发泡而形成发泡体的方式。作为发泡剂,可使用在发泡成型中通常使用的发泡剂,从环境保护及对于被发泡体的低污染性的观点考虑,优选使用高压的不活泼气体。
122.作为不活泼气体,只要对于树脂组合物无活性且能够浸渗,就可以采用任意适当的不活泼气体。作为这样的不活泼气体,可列举例如:二氧化碳、氮气、空气等。这些气体也可以混合使用。这些当中,从对于树脂材料(聚合物)的浸渗量多、浸渗速度快的观点考虑,优选二氧化碳。
123.不活泼气体优选为超临界状态。即,特别优选使用超临界状态的二氧化碳。在超临界状态下,不活泼气体对于树脂组合物的溶解度进一步增大,能够以高浓度混入不活泼气体,并且压力急剧下降时,不活泼气体成为高浓度,因此,气泡核的产生变多,即使气孔率相同,该气泡核成长而能够形成的气泡的密度与其它状态的情况相比也更大,因此,能够得到微细的气泡。需要说明的是,二氧化碳的临界温度为31℃、临界压力为7.4mpa。
124.作为通过在树脂组合物中浸渗高压的不活泼气体而形成发泡体的方法,可列举例如经过下述工序而形成的方法等:在高压下使树脂组合物中浸渗不活泼气体的气体浸渗工序;在该工序后降低压力而使树脂发泡的减压工序;以及根据需要进行的通过加热而使气泡生长的加热工序。在该情况下,可以使预先成型的未发泡成型体浸于不活泼气体中,另外也可以在加压状态下使不活泼气体浸渗于熔融的树脂组合物中之后在减压时实施成型。这些工序可以以间歇方式、连续方式中的任意方式进行。即,可以是预先将树脂组合物成型为片状等适宜的形状并制成未发泡树脂成型体后、在该未发泡树脂成型体中浸渗高压的气体并释放压力而发泡的间歇方式;也可以是在加压下将树脂组合物与高压的气体一起进行混炼、成型,同时释放压力而同时进行成型和发泡的连续方式。
125.以下示出以间歇方式制造发泡体的例子。例如,通过使用单螺杆挤出机、双螺杆挤出机等挤出机对树脂组合物进行挤出而制作发泡体成型用树脂片。或者使用辊、凸轮、捏合机、班伯里型等设置有叶片的混炼机预先对树脂组合物进行均匀混炼,再利用热板的加压等压制加工成给定的厚度从而制作未发泡树脂成型体。将这样得到的未发泡树脂成型体放入高压容器中,注入高压不活泼气体(超临界状态的二氧化碳等),在未发泡树脂成型体中浸渗不活泼气体。在充分地浸渗有不活泼气体的时刻,释放压力(通常至大气压),在树脂中产生气泡核。可以使气泡核直接在室温下成长,但根据情况,也可以通过加热使其成长。作为加热的方法,可采用水浴、油浴、热辊、热风烘箱、远红外线、近红外线、微波等公知、惯用
的方法。如此地使气泡生长之后,通过冷水等急剧冷却并将形状固定化,由此可以得到发泡体。需要说明的是,供于发泡的未发泡树脂成型体不限于片状物,也可以根据用途使用各种形状的未发泡树脂成型体。另外,供于发泡的未发泡树脂成型体除了挤出成型、加压成型以外,也可以通过注塑成型等其它成型法来制作。
126.以下示出以连续方式制造发泡体的例子。例如,通过下述混炼浸渗工序、成型减压工序进行发泡成型,该混炼浸渗工序一边使用单螺杆挤出机、双螺杆挤出机等挤出机对树脂组合物进行混炼,一边注入(导入)高压的气体(特别是不活泼气体、进而为二氧化碳),使高压的气体充分地浸渗于树脂组合物中;该成型减压工序通过设置于挤出机的前端的模头等将树脂组合物挤出,由此释放压力(通常至大气压),同时进行成型和发泡。另外,以连续方式进行发泡成型时,可以根据需要设置通过加热而使气泡成长的加热工序。如此地使气泡成长之后,可以根据需要通过冷水等急剧地进行冷却将形状固定化。另外,高压的气体的导入可以连续地进行,也可以不连续地进行。此外,在混炼浸渗工序及成型减压工序中,可以使用例如挤出机、注塑成型机。需要说明的是,作为使气泡核成长时的加热方法,可列举水浴、油浴、热辊、热风烘箱、远红外线、近红外线、微波等任意适当的方法。作为发泡体的形状,可采用任意适当的形状。作为这样的形状,可列举例如:片状、棱柱状、圆筒状、异型形状等。
127.从能够得到高发泡的发泡体的方面考虑,将树脂组合物发泡成型时的气体的混合量例如相对于树脂组合物100重量份优选为2重量份~10重量份、更优选为2.5重量份~8重量份、进一步优选为3重量份~6重量份。
128.使不活泼气体在树脂组合物中浸渗时的压力可以考虑操作性等适宜选择。这样的压力例如优选为6mpa以上(例如为6mpa~100mpa)、更优选为8mpa以上(例如为8mpa~50mpa)。需要说明的是,从保持二氧化碳的超临界状态的观点考虑,在使用超临界状态的二氧化碳的情况下的压力优选为7.4mpa以上。在压力低于6mpa的情况下,发泡时的气泡成长显著,气泡直径变得过大,有时无法得到优选的平均气孔直径(平均气泡直径)。这是因为,压力低时,气体的浸渗量与高压时相比相对较少,气泡核形成速度降低,形成的气泡核数变少,所以平均每1个气泡的气体量反而增加,气泡直径变得极大。另外,在低于6mpa的压力区域内,浸渗压力仅仅稍有变化就会导致气泡直径、气泡密度大幅变化,因此,气泡直径及气泡密度的控制容易变得困难。
129.气体浸渗工序中的温度根据使用的不活泼气体、树脂组合物中的成分的种类等而不同,能够在宽范围内选择。在考虑了操作性等的情况下,优选为10℃~350℃。在未发泡成型体中浸渗不活泼气体时的浸渗温度在间歇式的情况下优选为10℃~250℃、更优选为40℃~230℃。另外,将浸渗有气体的熔融聚合物挤出并同时进行发泡和成型时的浸渗温度在连续式的情况下优选为60℃~350℃。需要说明的是,在使用二氧化碳作为不活泼气体的情况下,为了保持超临界状态,浸渗时的温度优选为32℃以上、更优选为40℃以上。
130.在减压工序中,作为减压速度,为了得到均匀的微细气泡,优选为5mpa/秒~300mpa/秒。
131.加热工序中的加热温度优选为40℃~250℃、更优选为60℃~250℃。
132.《《2.发泡构件》》
133.本发明的发泡构件具备:由上述树脂发泡体构成的树脂发泡层、和配置于该树脂
发泡层的至少一侧的粘合剂层。
134.本发明的发泡构件所具有的树脂发泡层的厚度优选为30μm~5000μm、更优选为35μm~4000μm、进一步优选为40μm~3000μm、特别优选为45μm~2500μm。通过使树脂发泡层的厚度在上述范围内,该树脂发泡层即使对于微小间隙也能够容易地追随。另外,通过使树脂发泡层的厚度在上述范围内,能够均匀地含有气泡,能够展现出优异的冲击吸收性。
135.粘合剂层的厚度优选为5μm~300μm、更优选为6μm~200μm、进一步优选为7μm~100μm、特别优选为8μm~50μm。通过使粘合剂层的厚度在上述范围内,本发明的发泡构件能够发挥优异的冲击吸收性。
136.作为粘合剂层,可采用由任意适当的粘合剂形成的层。作为构成粘合剂层的粘合剂,可列举例如:橡胶类粘合剂(合成橡胶类粘合剂、天然橡胶类粘合剂等)、氨基甲酸酯类粘合剂、丙烯酸氨基甲酸酯类粘合剂、丙烯酸类粘合剂、有机硅类粘合剂、聚酯类粘合剂、聚酰胺类粘合剂、环氧类粘合剂、乙烯基烷基醚类粘合剂、氟类粘合剂、橡胶类粘合剂等。作为构成粘合剂层的粘合剂,优选为选自丙烯酸类粘合剂、有机硅类粘合剂、橡胶类粘合剂中的至少一种。这样的粘合剂可以仅为一种,也可以为两种以上。粘合剂层可以为一层,也可以为两层以上。
137.作为粘合剂,如果以粘合方式进行分类,则可列举例如:乳液型粘合剂、溶剂型粘合剂、紫外线交联型(uv交联型)粘合剂、电子束交联型(eb交联型)粘合剂、热熔融型粘合剂(热熔型粘合剂)等。这样的粘合剂可以仅为一种,也可以为两种以上。
138.粘合剂层的水蒸气透湿度优选为50(g/(m2·
24小时))以下、更优选为30(g/(m2·
24小时))以下、进一步优选为20(g/(m2·
24小时))以下、特别优选为10(g/(m2·
24小时))以下。如果粘合剂层的水蒸气透湿度在上述范围内,则本发明的发泡片能够使冲击吸收性稳定化而不会受到由水分导致的影响。
139.可以在不损害本发明效果的范围内在构成粘合剂层的粘合剂中包含任意适当的其它成分。
140.作为其它成分,可列举例如:其它聚合物成分、软化剂、防老化剂、固化剂、增塑剂、填充剂、抗氧剂、热聚合引发剂、光聚合引发剂、紫外线吸收剂、光稳定剂、着色剂(颜料、染料等)、溶剂(有机溶剂)、表面活性剂(例如,离子型表面活性剂、有机硅类表面活性剂、氟类表面活性剂等)、交联剂(例如多异氰酸酯类交联剂、有机硅类交联剂、环氧类交联剂、烷基醚化三聚氰胺类交联剂等)等。需要说明的是,热聚合引发剂、光聚合引发剂可以包含于用于形成聚合物成分的材料中。
141.本发明的发泡构件可以通过任意适当的方法来制造。本发明的发泡构件的制造方法例如可列举:将树脂发泡层与粘合剂层层叠而制造的方法;在将粘合剂层的形成材料与树脂发泡层层叠后通过固化反应等形成粘合剂层而制造的方法等。
142.实施例
143.以下,通过实施例对本发明进行具体说明,但本发明不受这些实施例的任何限定。需要说明的是,实施例等中的试验及评价方法如下所述。需要说明的是,在记载为“份”的情况下,只要没有特别说明,则是指“重量份”,在记载为“%”的情况下,只要没有特别说明,则是指“重量%”。
144.<表观密度的测定方法>
145.树脂发泡体的密度(表观密度)如下所述地进行计算。将实施例/比较例中得到的树脂发泡结构体冲裁成20mm
×
20mm尺寸,作为试验片,用卡尺测定试验片的尺寸,接下来,通过电子天平测定试验片的重量,并且通过下式进行计算。
146.表观密度(g/cm3)=试验片的重量/试验片的体积
147.<50%压缩载荷的测定方法>
148.按照jis k 6767中记载的发泡体的压缩硬度测定方法进行了测定。具体而言,将实施例/比较例中得到的树脂发泡结构体切出30mm
×
30mm尺寸,作为试验片,以压缩速度10mm/min压缩至压缩率成为50%为止,将此时的应力(n)换算成平均每单位面积(1cm2)的值,作为50%压缩载荷(n/cm2)。
149.<平均气泡直径(平均气孔直径)、气泡直径(气孔直径)的变动系数的测定方法>
150.作为计测器,使用数字显微镜(商品名“vhx
‑
500”,、keyence株式会社制),导入实施例/比较例中得到的树脂发泡结构体的气泡部的放大图像,使用相同计测器的解析软件,通过进行图像解析而求出平均气泡直径(平均气孔直径)(μm)。需要说明的是,导入的放大图像的气泡数为400个左右。另外,根据气孔直径的全部数据计算标准偏差,使用下式计算出变动系数。
151.变动系数=标准偏差/平均气泡直径(平均气孔直径)
152.<气泡率(气孔率)的测定方法>
153.在温度23℃、湿度50%的环境中进行了测定。通过100mm
×
100mm的冲裁刀模对实施例/比较例中得到的树脂发泡结构体进行冲裁,测定了冲裁后的试样的尺寸。另外,用测定端子的直径(φ)为20mm的1/100千分尺测定了厚度。根据这些值计算出实施例/比较例中得到的树脂发泡结构体的体积。接下来,通过最小刻度0.01g以上的托盘天平测定了实施例/比较例中得到的树脂发泡结构体的重量。根据这些值计算出实施例/比较例中得到的树脂发泡结构体的气泡率(气孔率)。
154.<树脂发泡体在650℃下的残渣的测定方法>
155.在铂制容器中加入5mg的实施例/比较例中得到的树脂发泡结构体,在氮气气氛中以升温速度20℃/min的条件在25℃~680℃的测定范围内进行升温,使用tg/dta6200(sii nanotechnology公司制)测定了在650℃下的残渣。
156.<树脂发泡体的拉伸模量的测定方法>
157.基于jis k 6767的拉伸伸长率一项,测定发泡体的拉伸伸长率(%)、拉伸强度,在以拉伸伸长率为x轴、拉伸强度为y轴的图表中,计算出拉伸伸长率0%~10%的区域中的拉伸强度的变化的比例作为拉伸模量。
158.<树脂发泡体的应力保持力的测定方法>
159.将树脂发泡体(宽10mm
×
长100mm)沿着长度方向以300m/min的速度拉伸20%,求出刚拉伸后的拉伸强度、和拉伸后保持了120秒钟后的拉伸强度的比率(保持120秒钟后的拉伸强度/刚拉伸后的应力保持力
×
100),将该比率作为树脂发泡体的应力保持力。
160.<应力分散度(性)的测定方法>
161.图1是应力分散度的测定中使用的应力缓和试验机1000的剖面示意图。
162.如图1所示,在铁制的支撑体100上放置聚碳酸酯板(200mm
×
300mm
×
厚度1mm)200,在其上放置应力测定膜300(商品名“prescale”(两片,微压用(4lw),富士较胶片株式
会社制,具有加压的部分显色的面的片,50mm
×
50mm
×
厚度0.16mm)。接下来,在应力测定膜300上放置测定对象的实施例/比较例中得到的树脂发泡结构体(150mm
×
200mm
×
厚度0.5mm)400,在其上粘贴双面胶粘带(no.5603、日东电工制、厚度0.03mm)500,配置厚度0.3mm的间隔件600,在最上部放置abs板(200mm
×
300mm
×
厚度3mm)700。从其上在中心部放置铁球(φ25mm)800,施加了100n的负载1min。
163.然后,观察应力测定膜300的颜色的变化,将颜色不从应力测定膜300的中心展开而成为点状者设为c、将颜色从应力测定膜300的中心起展开至25mm者设为b、将颜色从应力测定膜300的中心起大幅展开至50mm端部者设为a。
164.<尺寸变化率的测定方法>
165.通过jis k 6767:1999k“发泡塑料
‑
聚乙烯
‑
试验方法”中的b法对树脂发泡体的尺寸变化率进行了测定。
166.将试验片尺寸设为150mm
×
150mm。在试验片的中央部以50mm间隔在纵向及横向上分别标记相互平行的3条直线。接着将该试验片投入120℃的热风循环式干燥机,放置了500小时。然后,将试验片取出,在常温下放置了1小时后,测定标记线的长度。根据加热前的标记线的平均长度l0(mm)、加热后的标记线的平均长度l1(mm)并通过式|(l1-l0)|/l0
×
100求出了尺寸变化率。
167.〔实施例1〕
168.使用株式会社日本制钢所(jsw)制造的双螺杆混炼机将聚丙烯[熔体流动速率(mfr)(230℃):0.40g/10min]:50重量份、聚丙烯[熔体流动速率(mfr)(230℃):2.40g/10min]:25重量份、聚烯烃类弹性体[商品名“thermorun 5850n”、三菱化学制]:35重量份、氢氧化镁:100重量份(商品名“kisuma 5p”协和化学工业制)、碳(商品名“旭#35”、旭碳株式会社制):10重量份、及硬脂酸单甘油酯:1重量份在200℃的温度下混炼后,以线料状挤出,水冷后成型为颗粒状。将该颗粒投入株式会社日本制钢所制造的单螺杆挤出机,在220℃的气氛中以13(注入后12)mpa的压力注入了二氧化碳气体。相对于树脂100重量份,以3.5重量份的比例注入了二氧化碳气体。使二氧化碳气体充分饱和后,冷却至适于发泡的温度后,从模头挤出,得到了厚度为1.8mm的片状的树脂发泡体a。
[0169]
对于该发泡体而言,其表观密度为0.085g/cm3、650℃残渣为36%、拉伸模量为1.6mpa、应力保持率为70%。将包括这些结果在内的对于树脂发泡体a的评价结果示于表1。
[0170]
〔实施例2〕
[0171]
与实施例1同样地得到了树脂发泡体a。使上述树脂发泡体a通过一个辊加热至200℃的一对辊的辊间(辊与辊之间的间隙),得到了厚度为0.15mm的树脂发泡体b。需要说明的是,辊间的空隙(间隙)按照可得到厚度为0.15mm的树脂发泡体b的方式设定。
[0172]
对于该发泡体而言,其表观密度为0.18g/cm3、650℃残渣为36%、拉伸模量为2.1mpa、应力保持率为66%。将包括这些结果在内的对于树脂发泡体b的评价结果示于表1。
[0173]
〔实施例3〕
[0174]
使用株式会社日本制钢所(jsw)制造的双螺杆混炼机将聚丙烯[熔体流动速率(mfr)(230℃):0.40g/10min]:19重量份、聚丙烯[熔体流动速率(mfr)(230℃):1.1g/10min]:19重量份、聚烯烃类弹性体[熔体流动速率(mfr):6g/10min、jis a硬度:79
°
]:67重量份、氢氧化镁:80重量份(商品名“kisuma5p”协和化学工业制)、碳(商品名“旭#35”旭碳株
式会社制):10重量份、及硬脂酸单甘油酯:1重量份在200℃的温度下混炼后,以线料状挤出,水冷后成型为颗粒状。将该颗粒投入株式会社日本制钢所制造的单螺杆挤出机,在220℃的气氛中以13(注入后12)mpa的压力注入了二氧化碳气体。相对于树脂100重量份,以3重量份的比例注入了二氧化碳气体。使二氧化碳气体充分饱和后,冷却至适于发泡的温度后,从模头挤出,得到了厚度为1.8mm的片状的树脂发泡体c。
[0175]
对于该发泡体而言,其表观密度为0.07g/cm3、650℃残渣为34%、拉伸模量为0.6mpa、应力保持率为75%。将包括这些结果在内的对于树脂发泡体c的评价结果示于表1。
[0176]
〔实施例4〕
[0177]
使用株式会社日本制钢所(jsw)制造的双螺杆混炼机将聚丙烯[熔体流动速率(mfr)(230℃):0.40g/10min]:32.5重量份、聚丙烯[熔体流动速率(mfr)(230℃):1.1g/10min]:32.5重量份、聚烯烃类弹性体[熔体流动速率(mfr):6g/10min、jis a硬度:79
°
]:35重量份、氢氧化镁:120重量份(商品名“kisuma 5p”协和化学工业制)、碳(商品名“旭#35”旭碳株式会社制):10重量份、及硬脂酸单甘油酯:1重量份在200℃的温度下混炼后,以线料状挤出,水冷后成型为颗粒状。将该颗粒投入株式会社日本制钢所制造的单螺杆挤出机,在220℃的气氛中以13(注入后12)mpa的压力注入二氧化碳气体。相对于树脂100重量份,以3.5重量份的比例注入了二氧化碳气体。使二氧化碳气体充分饱和后,冷却至适于发泡的温度后,从模头挤出,得到了厚度为2.0mm的片状的树脂发泡体d。
[0178]
对于该发泡体而言,其表观密度为0.07g/cm3、650℃残渣为45%、拉伸模量为0.71mpa、应力保持率为63%。包括这些结果在内,将关于树脂发泡体d的评价结果示于表1。
[0179]
〔实施例5〕
[0180]
准备了表观密度为0.3g/cm3、650℃残渣为10%、拉伸模量为2.4mpa、应力保持率为65%的以聚乙烯为主成分的树脂发泡体e。将关于树脂发泡体e的评价结果示于表1。
[0181]
〔比较例1〕
[0182]
使用株式会社日本制钢所(jsw)制造的双螺杆混炼机将聚丙烯[熔体流动速率(mfr)(230℃):0.40g/10min]:22.5重量份、聚丙烯[熔体流动速率(mfr)(230℃):1.1g/10min]:22.5重量份、聚烯烃类弹性体[熔体流动速率(mfr):6g/10min、jis a硬度:79
°
]:55重量份、氢氧化镁:10重量份(商品名“kisuma 5p”协和化学工业制)、碳(商品名“旭#35”旭碳株式会社制):10重量份、及硬脂酸单甘油酯:1重量份在200℃的温度下混炼后,以线料状挤出,水冷后成型为颗粒状。将该颗粒投入株式会社日本制钢所制造的单螺杆挤出机,在220℃的气氛中以13(注入后12)mpa的压力注入了二氧化碳气体。相对于树脂100重量份,以5.5重量份的比例注入了二氧化碳气体。使二氧化碳气体充分饱和后,冷却至适于发泡的温度后,从模头挤出,得到了厚度为1.8mm的片状的树脂发泡结构体。使上述树脂发泡体通过一个辊加热至200℃的一对辊的辊间(辊与辊之间的间隙),得到了厚度为0.15mm的树脂发泡体f。需要说明的是,辊间的空隙(间隙)按照可得到厚度为0.15mm的树脂发泡体f的方式设定。
[0183]
对于该发泡体而言,其表观密度为0.07g/cm3、650℃残渣为14%、拉伸模量为0.55mpa、应力保持率为56%。将包括这些结果在内的对于树脂发泡体f的评价结果示于表1。
[0184]
[表1]
[0185] 实施例1实施例2实施例3实施例4实施例5比较例1表观密度[g/cm3]0.0850.180.070.070.30.0750%压缩载荷[n/cm2]9133.56203.5拉伸弹性模量[mpa]1.62.10.60.712.40.55应力保持率[%]706675636556r:发泡体的650℃残渣[wt%]363634451014(100
‑
r)/d
÷
1007.533.569.437.863.0012.29气泡直径[μm]807090709075气泡直径变动系数0.30.320.40.40.40.4气泡率[%]91.58293937093120℃尺寸变化率[%]0.80.30.90.80.35应力分散性[%]aaaabb
[0186]
根据120℃尺寸变化率的结果可知,本发明的树脂组合物的耐热性优异。另外,本发明的树脂组合物除了耐热性优异以外,应力分散性也优异。
[0187]
工业实用性
[0188]
本发明的树脂发泡体例如可以适宜地用作电子设备用的缓冲材料。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。