1.本发明涉及一种双马来酰亚胺化合物、使用其的感光性树脂组成物、其硬化物及半导体组件。本发明的感光性树脂组成物可适用于半导体组件用保护膜、层间绝缘膜、再配线层的绝缘膜等。
背景技术:
2.已知半导体组件用保护膜、形成于半导体表层的层间绝缘膜和再配线层的绝缘膜使用耐热性、电特性及机械特性优异的含有聚酰亚胺前驱物或聚苯并噁唑前驱物的感光性树脂组成物。作为上述含有聚酰亚胺前驱物的感光性树脂组成物,例如日本特开昭54
‑
109828号公报(专利文献1)中记载有含有聚酰胺酸、具有聚合性不饱和键的化合物及光聚合引发剂的树脂组成物。另外,日本特开2008
‑
83468号公报(专利文献2)中记载有含有聚酰胺酸酯组成物及光聚合引发剂的树脂组成物。在此种树脂组成物中获得的感光性聚酰亚胺前驱物为通过利用光聚合引发剂使不饱和键进行光交联而获得图案的负性感光性材料。另外,作为上述含有聚苯并噁唑前驱物的感光性树脂组成物,例如日本特开昭56
‑
27140号公报(专利文献3)及日本特开平11
‑
237736号公报(专利文献4)中记载有含有聚苯并噁唑前驱物及醌二叠氮化合物的树脂组成物。此种树脂组成物为以下正性感光性材料,其通过利用光照射使醌二叠氮变成茚羧酸,而经光照射的部分(曝光部)溶解于碱性显影液,从而获得图案。
3.如专利文献1~4中记载的聚酰亚胺前驱物或聚苯并噁唑前驱物的硬化反应中需进行脱水闭环反应,因此为了使其硬化,必须加热到至少超过230℃。然而,在加热温度如此高的情形下,存在对半导体组件造成损害的可能性,并且由于硅晶圆等基板与由感光性树脂组成物所构成的膜之间的线热膨胀系数不同,因此存在冷却至室温前的温度差导致硬化后的膜内产生残留应力的问题。进而,在如专利文献1~4中记载的感光性树脂组成物中,为了提高其耐热性或机械物性,而使硬化所得的树脂的聚合物骨架成为由刚性芳香族化合物构成的骨架,因此,存在硬化后的拉伸弹性模量变高、与被粘着体的密接性降低或残留应力变得更大的问题。此种残留应力导致硅晶圆等基板的翘曲,成为引起倒装晶圆封装等中与插入物的接合可靠性降低、半导体制造步骤中硅晶圆等基板的操作性降低等异常的原因。尤其近年来,从半导体组件的小型化、薄型化的观点出发而推进硅晶圆的薄型化,或从提高量产性的观点出发而推进硅晶圆的大口径化(量产水平为300mm直径左右,将来为450mm直径左右),伴随于此,关于此种残留应力的问题变得更重大。
4.另外,作为用以降低进行硬化的温度(硬化温度)的感光性树脂组成物,日本特开2009
‑
258433号公报(专利文献5)及日本特开2009
‑
175356号公报(专利文献6)中记载有含有聚苯并噁唑前驱物的树脂组成物。进而,作为其它感光性树脂组成物,例如日本特开2010
‑
256532号公报(专利文献7)中记载有一种感光性树脂组成物,其含有利用由二聚酸衍生的胺化合物及二胺与四羧酸二酐的缩合反应所获得的聚酰胺酸。另外,日本特表2006
‑
526014号公报(专利文献8)中记载有一种预先使酰胺酸结构闭环并引入马来酰亚胺基作为聚合性官能团而形成的聚马来酰亚胺化合物,含有该聚马来酰亚胺化合物的感光性树脂组成物在美国申请案公开第2011/0049731号说明书(专利文献9)中有记载。
5.背景技术文献
6.专利文献
7.专利文献1:日本特开昭54
‑
109828号公报
8.专利文献2:日本特开2008
‑
83468号公报
9.专利文献3:日本特开昭56
‑
27140号公报
10.专利文献4:日本特开平11
‑
237736号公报
11.专利文献5:日本特开2009
‑
258433号公报
12.专利文献6:日本特开2009
‑
175356号公报
13.专利文献7:日本特开2010
‑
256532号公报
14.专利文献8:日本特表2006
‑
526014号公报
15.专利文献9:美国申请案公开第2011/0049731号说明书
技术实现要素:
16.[本发明旨在解决的技术问题]
[0017]
本发明人等发现,聚酰亚胺前驱物或聚苯并噁唑前驱物在436nm及365nm时的吸收较大,因此在使用于半导体的保护膜等的制造步骤中标准地使用的缩小投影曝光机(步进机;光源波长:365nm、436nm)的情形下,随着膜厚变厚,到达至底部的光减少,而难以形成图案。尤其是半导体组件用保护膜等的膜厚一般为5μm以下,但配线引起的凹凸导致实际上存在膜厚为10μm以上的部分的情况较多,此种部分存在无法充分发挥图案化性能,而晶圆设计受到限制的问题。
[0018]
另外,专利文献7中记载的聚酰胺酸具有由自二聚酸衍生的胺化合物(二聚二胺)及四羧酸二酐获得的聚酰胺酸结构,可期待获得可挠性优异的硬化物。然而,专利文献7中记载的聚酰胺酸不具有光聚合性官能团,因此必须向树脂组成物添加光聚合性化合物,例如,若将作为光聚合性化合物的一般的丙烯酸化合物等具有多个聚合性官能团的多官能聚合性化合物与上述聚酰胺酸一起使用,则存在利用光聚合的交联反应进行而硬化后的拉伸弹性模量变高的问题。进而,专利文献7中记载的感光性树脂组成物在硬化反应中必须进行酰胺酸结构的脱水闭环反应,因此,必须以超过230℃的高温进行加热,而也存在产生导致硅晶圆等基板的翘曲的残留应力的问题。
[0019]
进而,专利文献8中记载的聚马来酰亚胺化合物为可溶性酰亚胺低聚物,因此含有其的专利文献9中记载的树脂组成物能够以相对较低的温度进行硬化。然而,若使用专利文献9中记载的树脂组成物,则存在与形成在硅晶圆或晶圆上的sin膜或sio2膜等无机表面保护膜(钝化膜)或者导电性金属配线材料(铜等)的密接性明显降低的问题、或者难以形成微细图案的问题。另外,作为提高密接性的方法,可列举通过增加曝光量而提高利用光聚合的交联反应的效率的方法,但专利文献8中记载的聚马来酰亚胺化合物与通常用作光聚合性化合物的丙烯酸化合物等相比较需要非常多的曝光量,因此存在在半导体制造步骤中导致生产性降低的问题。进而,作为降低硬化后的膜内的残留应力、或提高图案化性能的方法,
可列举使膜厚变薄的方法,但若使膜厚变薄,则存在损害本来作为半导体组件用保护膜或绝缘膜的绝缘性的问题。
[0020]
本发明是鉴于上述已知技术所具有的课题而完成的,其目的在于:提供一种双马来酰亚胺化合物、使用其的感光性树脂组成物、其硬化物及具备该硬化物的半导体组件;上述双马来酰亚胺化合物能够以相对较低的曝光量形成微细的图案,且无需进行如已知的高温下的热硬化,而可获得拉伸弹性模量充分小且与无机表面保护膜或金属配线材料的密接性优异的硬化物。
[0021]
[解决技术问题的技术手段]
[0022]
本发明人等为了达成上述目的,反复进行了努力研究,结果发现,通过使用包含特定的双马来酰亚胺化合物(i)的感光性树脂组成物,能够以相对较低的曝光量形成微细的图案,另外,无需热硬化,或者即便在视需要进行热硬化的情形下,也无需进行如已知的高温下的热硬化。进而发现,通过使用此种感光性树脂组成物所获得的硬化物的拉伸弹性模量充分小,与无机表面保护膜或金属配线材料的密接性优异,可尤其适宜地用作例如必须保持高绝缘性的半导体组件的表面保护膜、层间绝缘膜及再配线层的绝缘膜等,从而完成了本发明。
[0023]
即,本发明涉及以下内容:
[0024]
1.一种双马来酰亚胺化合物(i),其具有环状酰亚胺键,且是使由二聚酸衍生的二胺(a)、具有脂环结构的四羧酸二酐(c)及马来酸酐反应而获得的。
[0025]
2.如1中记载的双马来酰亚胺化合物(i),其是使所述二胺(a)、所述四羧酸二酐(c)、所述马来酸酐以及进一步由所述二聚酸衍生的所述二胺(a)以外的有机二胺(b)反应而获得的。
[0026]
3.如1或2中记载的双马来酰亚胺化合物(i),所述双马来酰亚胺化合物(i)以下述通式(1)表示:
[0027]
[化学式1]
[0028][0029]
[在式(1)中,r1表示源自二聚酸的二价烃基(a),r2表示源自二聚酸的二价烃基(a)以外的二价有机基团(b),r3表示选自由源自二聚酸的二价烃基(a)及源自二聚酸的二价烃基(a)以外的二价有机基团(b)所组成的群中的任一种,r4及r5分别独立地表示选自具有单环式或缩合多环式脂环结构的碳数为4~40的四价有机基团、具有单环式脂环结构的有机基团直接或经由交联结构互相连接的碳数为8~40的四价有机基团、及具有包含脂环结构与芳香环两者的半脂环结构的碳数为8~40的四价有机基团中的一种以上的有机基团;m为1~30的整数,n为0~30的整数,r4及r5可分别相同,也可不同]。
[0030]
4.如1至3中任一项所记载的双马来酰亚胺化合物(i),其中,所述四羧酸二酐(c)以通式(2)表示:
[0031]
[化学式2]
[0032][0033]
(在式中,cy为包含烃环的碳数为4~40的四价有机基团,该有机基也可包含芳香族环。)
[0034]
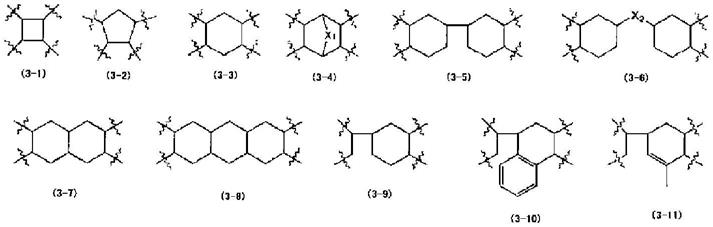
5.如4中记载的双马来酰亚胺化合物,其中,所述cy选自由式(3
‑
1)~(3
‑
11)所组成的群:
[0035]
[化学式3]
[0036][0037]
(在通式(3
‑
4)中,x1为直接键结、氧原子、硫原子、磺酰基或碳数为1~3的二价有机基团;在通式(3
‑
6)中,x2为直接键结、氧原子、硫原子、磺酰基、碳数为1~3的二价有机基团或亚芳基);
[0038]
6.如1至4中任一项所记载的双马来酰亚胺化合物(i),其中所述四羧酸二酐(c)选自以下化合物中的一种以上:
[0039]
1,2,3,4
‑
环丁烷四羧酸二酐(cbda)、1,2
‑
二甲基
‑
1,2,3,4
‑
环丁烷四羧酸二酐、1,2,3,4
‑
四甲基
‑
1,2,3,4
‑
环丁烷四羧酸二酐、1,2,3,4
‑
环戊烷四羧酸二酐、1,2,4,5
‑
环己烷四羧酸二酐(h
‑
pmda)、1,1
’‑
双环己烷
‑
3,3’,4,4
’‑
四羧酸
‑
3,4:3’,4
’‑
二酐(h
‑
bpda)、4
‑
(2,5
‑
二氧四氢呋喃
‑3‑
基)
‑
1,2,3,4
‑
四氢化萘
‑
1,2
‑
二羧酸酐、5
‑
(2,5
‑
二氧四氢呋喃基)
‑3‑
甲基
‑3‑
环己烯
‑
1,2
‑
二羧酸二酐、双环[2.2.2]辛
‑7‑
烯
‑
2,3,5,6
‑
四羧酸二酐、2,3,4,5
‑
四氢呋喃四羧酸二酐、3,5,6
‑
三羧基
‑2‑
降莰烷乙酸二酐;
[0040]
7.如1至6中任一项所记载的双马来酰亚胺化合物(i),其中,所述四羧酸二酐(c)为下述式(4)的化合物:
[0041]
[化学式4]
[0042][0043]
8.如1至6中任一项所记载的双马来酰亚胺化合物(i),其中,所述四羧酸二酐(c)为下述式(5)的化合物:
[0044]
[化学式5]
[0045][0046]
9.如1至6中任一项所记载的双马来酰亚胺化合物(i),其中,所述四羧酸二酐(c)为下述式(6)的化合物:
[0047]
[化学式6]
[0048][0049]
10.如1至6中任一项所记载的双马来酰亚胺化合物(i),其中,所述四羧酸二酐(c)为下述式(7)的化合物:
[0050]
[化学式7]
[0051][0052]
11.一种感光性树脂组成物,其包含1至10中任一项所记载的双马来酰亚胺化合物(i)及光聚合引发剂(ii),且光聚合引发剂(ii)为具有肟结构或噻唑酮结构的化合物;
[0053]
12.如11中记载的感光性树脂组成物,其中,相对于100质量份的所述双马来酰亚胺化合物(i),所述光聚合引发剂(ii)的含量为0.1~15质量份;
[0054]
13.一种硬化物,其是使11或12中记载的感光性树脂组成物进行光硬化或光热硬化而获得的;
[0055]
14.一种半导体组件,其具备13中记载的硬化物作为选自由表面保护膜、层间绝缘膜及再配线层的绝缘膜所组成的群中的至少一种。
[0056]
[本发明的技术效果]
[0057]
根据本发明,能够提供一种双马来酰亚胺化合物、使用其的感光性树脂组成物、其硬化物及具备该硬化物的半导体组件;上述双马来酰亚胺化合物能够以较低的曝光量形成微细的图案,且无需进行如已知的高温下的热硬化,即可获得拉伸弹性模量充分小、且与无机表面保护膜或金属配线材料密接性优异的硬化物。
具体实施方式
[0058]
以下,根据本发明的适宜的实施形态对其进行详细说明。
[0059]
<双马来酰亚胺化合物(i)>
[0060]
本发明的双马来酰亚胺化合物(i)为具有两个马来酰亚胺基团的化合物,具有源自二聚酸的二价烃基(a)及环状酰亚胺键。此种双马来酰亚胺化合物(i)可通过使由二聚酸
衍生的二胺(a)、具有脂环结构的四羧酸二酐(c)及马来酸酐反应而获得。
[0061]
上述源自二聚酸的二价烃基(a)是指自二聚酸所含的二羧酸去除两个羧基所获得的二价残基。在本发明中,此种源自二聚酸的二价烃基(a)可通过以下方法引入至双马来酰亚胺化合物中:使通过将二聚酸所含的二羧酸所具有的两个羧基取代成胺基而获得的二胺(a)、下述四羧酸二酐(c)及马来酸酐反应形成酰亚胺键。
[0062]
在本发明中,上述二聚酸优选为碳数为20~60的二羧酸。作为上述二聚酸的具体例,可列举使亚麻油酸、油酸、次亚麻油酸等不饱和羧酸的不饱和键二聚化,之后进行蒸馏精制所获得的二聚酸。上述具体例的二聚酸主要含有碳数为36个的二羧酸,通常分别以约5质量%为限度含有碳数54个的三羧酸及单羧酸。本发明的由二聚酸衍生的二胺(a)(以下,视情形称为源自二聚酸的二胺(a))通过将上述二聚酸所含的各二羧酸所具有的两个羧基取代成胺基而获得,通常为混合物。在本发明中,作为此种源自二聚酸的二胺(a),例如可列举含有[3,4
‑
双(1
‑
氨基庚基)
‑6‑
己基
‑5‑
(1
‑
辛烯基)]环己烷等的二胺、或通过将这些二胺进一步氢化而使不饱和键饱和的二胺。
[0063]
作为使用此种源自二聚酸的二胺(a)引入至双马来酰亚胺化合物中的本发明的源自二聚酸的二价烃基(a),优选为由上述源自二聚酸的二胺(a)去除两个氨基所获得的残基。另外,在使用上述源自二聚酸的二胺(a)获得本发明的双马来酰亚胺化合物(i)时,作为上述源自二聚酸的二胺(a),可单独使用一种,也可组合使用组成不同的两种以上。进而,作为此种源自二聚酸的二胺(a),例如也可使用“priamine1074”(croda japan株式会社制造)等市售品。
[0064]
在本发明中,四羧酸二酐(c)是指邻接于酸酐基团具有脂环结构,并具有如在反应后形成双马来酰亚胺化合物时酰亚胺环邻接部位成为脂环结构的结构。只要酰亚胺环邻接部位成为脂环结构,也可除此以外在该结构内含有芳香环。
[0065]
在本发明中,双马来酰亚胺化合物(i)优选为下述通式(1)。在通式(1)中,r4和r5为源自四羧酸二酐(c)的结构。
[0066]
[化学式8]
[0067][0068]
[在式(1)中,r1表示源自二聚酸的二价烃基(a),r2表示源自二聚酸的二价烃基(a)以外的二价有机基团(b),r3表示选自由源自二聚酸的二价烃基(a)及源自二聚酸的二价烃基(a)以外的二价有机基团(b)所组成的群中的任一种,r4和r5分别独立地表示选自具有单环式或缩合多环式脂环结构的碳数为4~40(优选碳数为6~40)的四价有机基团、具有单环式脂环结构的有机基团直接或经由交联结构互相连接的碳数为8~40的四价有机基团、及具有包含脂环结构及芳香环两者的半脂环结构的碳数为8~40的四价有机基团中的一种以上的有机基团;m为1~30的整数,n为0~30的整数,r4和r5可分别相同,也可不同。]
[0069]
在本发明中,四羧酸二酐(c)优选为下述通式(2)所表示的具有脂环结构的四羧酸二酐(c)。下述通式(2)所表示的具有脂环结构的四羧酸二酐(c)中邻接于酸酐基团具有脂环结构。
[0070]
[化学式9]
[0071][0072]
(在式中,cy为含有烃环的碳数为4~40的四价有机基团,该有机基也可包含芳香族环。)
[0073]
在本发明中,四羧酸二酐(c)优选为下述通式(3
‑
1)~(3
‑
11)所表示的具有脂环结构的四羧酸二酐(c)。式(3
‑
1)~(3
‑
11)所表示的四羧酸二酐(c)具有包含以下有机基团的结构:具有单环式或缩合多环式脂环结构的碳数为4~40(优选碳数为6~40)的四价有机基团、具有单环式脂环结构的有机基团直接或经由交联结构互相连接的碳数为8~40的四价有机基团、及具有包含脂环结构及芳香环两者的半脂环结构的碳数为8~40的四价有机基团。
[0074]
[化学式10]
[0075][0076]
(在通式(3
‑
4)中,x1为直接键结、氧原子、硫原子、磺酰基或碳数为1~3的二价有机基团;在通式(3
‑
6)中,x2为直接键结、氧原子、硫原子、磺酰基、碳数为1~3的二价有机基或亚芳基。)
[0077]
本发明中使用的四羧酸二酐(c)具有以下有机基团:具有单环式或缩合多环式脂环结构的碳数为4~40(优选碳数为6~40)的四价有机基团、具有单环式脂环结构的有机基团直接或经由交联结构互相连接的碳数为8~40的四价有机基团、及具有包含脂环结构及芳香环两者的半脂环结构的碳数为8~40的四价有机基团。作为具有脂环结构的四羧酸二酐(c),具体而言,可列举:如1,2,3,4
‑
环丁烷四羧酸二酐(cbda)、1,2
‑
二甲基
‑
1,2,3,4
‑
环丁烷四羧酸二酐、1,2,3,4
‑
四甲基
‑
1,2,3,4
‑
环丁烷四羧酸二酐、1,2,3,4
‑
环戊烷四羧酸二酐、1,2,4,5
‑
环己烷四羧酸二酐(h
‑
pmda)、1,1
’‑
双环己烷
‑
3,3’,4,4
’‑
四羧酸
‑
3,4:3’,4
’‑
二酐(h
‑
bpda)、4
‑
(2,5
‑
二氧四氢呋喃
‑3‑
基)
‑
1,2,3,4
‑
四氢化萘
‑
1,2
‑
二羧酸酐、5
‑
(2,5
‑
二氧四氢呋喃基)
‑3‑
甲基
‑3‑
环己烯
‑
1,2
‑
二羧酸二酐、双环[2.2.2]辛
‑7‑
烯
‑
2,3,5,6
‑
四羧酸二酐、2,3,4,5
‑
四氢呋喃四羧酸二酐、3,5,6
‑
三羧基
‑2‑
降莰烷乙酸二酐的脂环式四羧酸二酐或者其芳香族环被烷基或卤素原子取代的化合物;如1,3,3a,4,5,9b
‑
六氢
‑
5(四氢
‑
2,5
‑
二氧
‑3‑
呋喃基)萘并[1,2
‑
c]呋喃
‑
1,3
‑
二酮的半脂环式四羧酸二酐或者其芳香族环的氢原子被取代为烷基或卤素原子的化合物。
[0078]
另外,由本发明的感光性树脂组成物获得的图案优选为分辨率较高。分辨率意指在使用感光性树脂组成物形成图案时获得的最小尺寸,可形成越细小的图案,则分辨率越
高。
[0079]
在本发明中,四羧酸二酐(c)优选为下述通式(4)所表示的具有脂环结构的四羧酸二酐(c)。
[0080]
[化学式11]
[0081][0082]
在本发明中,四羧酸二酐(c)优选为下述通式(5)所表示的具有脂环结构的四羧酸二酐(c)。
[0083]
[化学式12]
[0084][0085]
在本发明中,四羧酸二酐(c)优选为下述通式(6)所表示的具有脂环结构的四羧酸二酐(c)。
[0086]
[化学式13]
[0087][0088]
在本发明中,四羧酸二酐(c)优选为下述通式(7)所表示的具有脂环结构的四羧酸二酐(c)。
[0089]
[化学式14]
[0090][0091]
通过使用适当量的四羧酸二酐(c)、由二聚酸衍生的二胺(a)及马来酸酐,可获得显影时无皱褶或显影残渣的高残膜率、高感度的感光性树脂组成物。
[0092]
在本发明中,除具有脂环结构的四羧酸二酐(c)以外,也可添加不具有脂环结构的酸二酐、或邻接于酸酐基团包含芳香环的酸二酐。酸二酐总量中,四羧酸二酐(c)的下限值优选为40摩尔%以上,更优选为80摩尔%以上,特别优选为90摩尔%以上。上限为100摩尔%以下即可。在酸二酐总量中的四羧酸二酐(c)的含量未达40摩尔%的情形下,存在聚光率较低而无法获得小图案的开口的倾向,因此存在所获得的图案的分辨率降低的担忧。
[0093]
作为上述四羧酸二酐(c)以外的邻接于酸酐基团包含芳香环的酸二酐,具体而言,
可列举:均苯四甲酸二酐、4,4
’‑
氧双邻苯二甲酸二酐、3,3’,4,4
’‑
联苯四羧酸二酐、2,3,3’,4
’‑
联苯四羧酸二酐、2,2’,3,3
’‑
联苯四羧酸二酐、3,3’,4,4
’‑
二苯甲酮四羧酸二酐、2,2’,3,3
’‑
二苯甲酮四羧酸二酐、2,2
‑
双(3,4
‑
二羧基苯基)丙烷二酐、2,2
‑
双(2,3
‑
二羧基苯基)丙烷二酐、1,1
‑
双(3,4
‑
二羧基苯基)乙烷二酐、1,1
‑
双(2,3
‑
二羧基苯基)乙烷二酐、双(3,4
‑
二羧基苯基)甲烷二酐、双(2,3
‑
二羧基苯基)甲烷二酐、1,2,5,6
‑
萘四羧酸二酐、2,3,6,7
‑
萘四羧酸二酐、2,3,5,6
‑
吡啶四羧酸二酐、3,4,9,10
‑
苝四羧酸二酐等芳香族四羧酸二酐、或双(3,4
‑
二羧基苯基)砜二酐、双(3,4
‑
二羧基苯基)醚二酐、2,2
‑
双(3,4
‑
二羧基苯基)六氟丙烷二酐或者这些化合物的芳香族环被烷基或卤素原子取代的化合物、及具有酰胺基的酸二酐等芳香族酸二酐。这些可与含有碳数为4~40的脂环结构或半脂环结构的酸二酐组合两种以上使用。
[0094]
进而,作为本发明的双马来酰亚胺化合物(i),也可为使上述源自二聚酸的二胺(a)、上述源自二聚酸的二胺(a)以外的有机二胺(b)、上述四羧酸二酐(c)以及上述马来酸酐反应而获得的双马来酰亚胺化合物。通过使上述源自二聚酸的二胺(a)以外的有机二胺(b)进行共聚合,能够视需要控制所要求的物性,如使所获得的硬化物的拉伸弹性模量进一步降低。
[0095]
上述源自二聚酸的二胺(a)以外的有机二胺(b)(以下,视情形仅称为有机二胺(b))是指本发明中的所述源自二聚酸的二胺(a)所含的二胺以外的二胺。作为此种有机二胺(b),并无特别限制,例如可列举:1,6
‑
六亚甲基二胺等脂肪族二胺;1,4
‑
二氨基环己烷、1,3
‑
双(氨基甲基)环己烷等脂环式二胺;4,4
’‑
二氨基二苯醚、3,4
’‑
二氨基二苯醚、1,4
‑
双(4
‑
氨基苯氧基)苯、1,3
‑
双(氨基甲基)苯、1,3
‑
双(4
‑
氨基苯氧基)苯、1,3
‑
双(3
‑
氨基苯氧基)苯、1,4
‑
二氨基苯、1,3
‑
二氨基苯、2,4
‑
二氨基甲苯、4,4
’‑
二氨基二苯基甲烷等芳香族二胺;4,4
’‑
二氨基二苯基砜;3,3
’‑
二氨基二苯基砜;4,4
‑
二氨基二苯甲酮;4,4
‑
二氨基二苯硫醚;2,2
‑
双[4
‑
(4
‑
氨基苯氧基)苯基]丙烷。在这些化合物中,就获得拉伸弹性模量更低的硬化物的观点而言,更优选为:1,6
‑
六亚甲基二胺等碳数为6~12个的脂肪族二胺;1,4
‑
二氨基环己烷等二氨基环己烷;2,2
‑
双[4
‑
(4
‑
氨基苯氧基)苯基]丙烷等芳香族骨架中具有碳数为1~4个的脂肪族结构的芳香族二胺。另外,在使用这些有机二胺(b)获得本发明的双马来酰亚胺化合物(i)时,可单独使用这些有机二胺(b)中的一种,也可组合两种以上使用。
[0096]
作为使上述源自二聚酸的二胺(a)、上述具有脂环结构的四羧酸二酐(c)及上述马来酸酐反应的方法或使上述源自二聚酸的二胺(a)、上述有机二胺(b)、上述具有脂环结构的四羧酸二酐(c)及上述马来酸酐反应的方法,并无特别限制,可适当采用公知的方法。例如,首先,将上述源自二聚酸的二胺(a)、上述四羧酸二酐(c)及视需要的上述有机二胺(b)在甲苯、二甲苯、四氢化萘、n,n
‑
二甲基乙酰胺、n
‑
甲基
‑2‑
吡咯啶酮等溶剂或这些溶剂的混合溶剂等溶剂中在室温(23℃左右)搅拌30~60分钟,由此合成聚酰胺酸,其次,向所获得的聚酰胺酸加入马来酸酐并在室温(23℃左右)搅拌30~60分钟,由此合成在两个末端加成有马来酸的聚酰胺酸。然后向该聚酰胺酸加入甲苯等与水共沸的溶剂,一边去除伴随酰亚胺化生成的水,一边在100~160℃的温度回流3~6小时,由此可获得作为目标的双马来酰亚胺化合物。另外,在此种方法中,也可进一步添加吡啶、甲磺酸等催化剂。
[0097]
作为上述反应中的原料的混合比,优选设为(源自二聚酸的二胺(a)所含的全部二胺及有机二胺(b)的总摩尔数):(具有脂环结构的四羧酸二酐(c)的总摩尔数 马来酸酐的
摩尔数的1/2)成为1:1。另外,在使用上述有机二胺(b)的情形下,就有表现出源自二聚酸的柔软性而获得弹性模量更低的硬化物的倾向的观点而言,(有机二胺(b)的摩尔数)/(源自二聚酸的二胺(a)所含的全部二胺的摩尔数)优选为1以下,更优选为0.4以下。此外,在使用上述有机二胺(b)的情形下,由源自二聚酸的二胺(a)及具有脂环结构的四羧酸二酐(c)所构成的酰胺酸单位与由有机二胺(b)及具有脂环结构的四羧酸二酐(c)所构成的酰胺酸单位的聚合形态可为无规聚合,也可为嵌段聚合。
[0098]
作为如此获得的双马来酰亚胺化合物(i),优选为下述通式(1)所表示的双马来酰亚胺化合物(i):
[0099]
[化学式15]
[0100][0101]
[在式(1)中,r1表示源自二聚酸的二价烃基(a),r2表示源自二聚酸的二价烃基(a)以外的二价有机基团(b),r3表示选自由源自二聚酸的二价烃基(a)及源自二聚酸的二价烃基(a)以外的二价有机基团(b)所组成的群中的任一种,r4和r5分别独立地表示选自具有单环式或缩合多环式脂环结构的碳数为4~40(优选碳数为6~40)的四价有机基团、具有单环式脂环结构的有机基团直接或经由交联结构互相连接的碳数为8~40的四价有机基团、及具有包含脂环结构及芳香环两者的半脂环结构的碳数为8~40的四价有机基团中的一种以上的有机基团;m为1~30的整数,n为0~30的整数,r4和r5可分别相同,也可不同。]
[0102]
作为上述式(1)中的上述源自二聚酸的二价烃基(a),如上所述。另外,在本发明中,上述式(1)中的源自二聚酸的二价烃基(a)以外的二价有机基团(b)是指自上述有机二胺(b)去除两个氨基所获得的二价残基。但是,在同一化合物中,上述源自二聚酸的二价烃基(a)与上述二价有机基团(b)并不相同。然后,上述式(1)中的上述四价有机基团是指由上述四羧酸二酐去除两个用
‑
co
‑
o
‑
co
‑
表示的基团所获得的四价残基。
[0103]
在上述式(1)中,m为包含上述源自二聚酸的二价烃基(a)的重复单位(以下,视情形称为源自二聚酸的结构)的数目,表示1~30的整数。在m的值超过上述上限的情形下,存在在溶剂中的溶解性降低、尤其是下述显影时在显影液中的溶解性降低的倾向。另外,作为m的值,就显影时在显影液中的溶解性适宜的观点而言,特别优选为3~10。
[0104]
在上述式(1)中,n为包含上述二价有机基(b)的重复单位(以下,视情形称为源自有机二胺的结构)的数目,表示0~30的整数。在n的值超过上述上限的情形下,存在获得的硬化物的柔软性变差而成为硬且脆的树脂的倾向。另外,作为n的值,就有可获得低弹性模量的硬化物的倾向的观点而言,特别优选为0~10。
[0105]
然后,在上述式(1)中的m为2以上的情形下,r1和r4在各重复单位间可相同,也可不同。另外,在上述式(1)中的n为2以上的情形下,r2和r5在各重复单位间可相同,也可不同。然后,作为上述式(1)所表示的双马来酰亚胺化合物,上述源自二聚酸的结构及上述源自有机二胺的结构可为无规,也可为嵌段。
[0106]
另外,在由上述源自二聚酸的二胺(a)、上述马来酸酐、上述四羧酸二酐(c)及视需要的上述有机二胺(b)获得本发明的双马来酰亚胺化合物(i)的情形下,在反应率为100%
时,上述n和m可通过上述源自二聚酸的二胺(a)所含的全部二胺、上述有机二胺(b)、上述马来酸酐及上述四羧酸二酐(c)的混合摩尔比表示。即,(m n):(m n 2)用(源自二聚酸的二胺(a)所含的全部二胺及有机二胺(b)的总摩尔数):(马来酸酐及四羧酸二酐(c)的总摩尔数)表示,m:n用(源自二聚酸的二胺(a)所含的全部二胺的摩尔数):(有机二胺(b)的摩尔数)表示,2:(m n)用(马来酸酐的摩尔数):(四羧酸二酐(c)的摩尔数)表示。
[0107]
然后,在本发明的双马来酰亚胺化合物(i)中,作为上述m与n之和(m n),就有获得弹性模量更低的硬化物的倾向的观点而言,优选为2~30。另外,作为上述m与n的比率(n/m),就表现出源自二聚酸的柔软性而获得弹性模量更低的硬化物的倾向的观点而言,优选为1以下,更优选为0.4以下。
[0108]
作为本发明的双马来酰亚胺化合物(i),可单独使用一种,也可组合两种以上使用。
[0109]
<光聚合引发剂(ii)>
[0110]
作为本发明的光聚合引发剂(ii),并无特别限制,可适当采用习知使用的,例如可列举:苯乙酮、2,2
‑
二甲氧基苯乙酮、对二甲氨基苯乙酮、米其勒酮、苯偶酰、安息香、安息香甲醚、安息香乙醚、安息香异丙醚、安息香正丙醚、安息香异丙醚、安息香正丁醚、苯偶酰二甲基缩酮、噻唑酮、2
‑
氯噻唑酮、2
‑
甲基噻唑酮、2,2
‑
二甲氧基
‑
1,2
‑
二苯基乙烷
‑1‑
酮、1
‑
羟基环己基苯基酮、2
‑
羟基
‑2‑
甲基
‑1‑
苯基丙烷
‑1‑
酮、1
‑
[4
‑
(2
‑
羟基乙氧基)
‑
苯基]
‑2‑
羟基
‑2‑
甲基
‑1‑
丙烷
‑1‑
酮、2
‑
羟基
‑1‑
{4
‑
[4
‑
(2
‑
羟基
‑2‑
甲基丙酰基)苄基]苯基}
‑2‑
甲基
‑
丙烷
‑1‑
酮、2
‑
甲基
‑1‑
(4
‑
甲基噻吩基)
‑2‑
吗啉基丙烷
‑1‑
酮、2
‑
苄基
‑2‑
二甲氨基
‑1‑
(4
‑
吗啉基苯基)
‑1‑
丁酮、2,4,6
‑
三甲基苯甲酰基二苯基氧化膦、双(2,4,6
‑
三甲基苯甲酰基)苯基氧化膦、1,2
‑
辛二酮、1
‑
[4
‑
(苯硫基)
‑2‑
(o
‑
苯甲酰肟)]、乙酮,1
‑
[9
‑
乙基
‑6‑
(2
‑
甲基苯甲酰基)
‑
9h
‑
咔唑
‑3‑
基]
‑
,1
‑
(o
‑
乙酰肟)、2,4
‑
二甲基噻唑酮等光聚合引发剂。作为此种光聚合引发剂(ii),可单独使用一种,也可组合两种以上使用。
[0111]
在这些化合物中,作为本发明的光聚合引发剂(ii),就可使用在半导体的保护膜等的制造步骤中标准地使用的缩小投影曝光机(步进机;光源波长:365nm、436nm)形成微细图案的观点而言,优选为使用可在曝光波长310~436nm(更优选为365nm)高效率地产生自由基的光聚合引发剂(ii)。另外,马来酰亚胺基团通常不进行利用自由基的均聚,而主要通过与由光聚合引发剂产生的自由基的反应进行双马来酰亚胺化合物的二聚反应,从而形成交联结构。因此,本发明人等推测,双马来酰亚胺化合物与通常用作光聚合性化合物的丙烯酸化合物等相比较,在表面上的反应性不足。因此,就可更有效率地产生自由基,曝光波长310~436nm(更优选为365nm)的反应性变高的观点而言,作为本发明的光聚合引发剂(ii),更优选为具有肟结构或噻唑酮结构的化合物。
[0112]
作为此种光聚合引发剂(ii),例如可列举:具有肟结构的1,2
‑
辛二酮、1
‑
[4
‑
(苯硫基)
‑
,2
‑
(o
‑
苯甲酰肟)](basf japan制造的“irgacure oxe
‑
01”)、乙酮,1
‑
[9
‑
乙基
‑6‑
(2
‑
甲基苯甲酰基)
‑
9h
‑
咔唑
‑3‑
基]
‑
,1
‑
(o
‑
乙酰肟)(basf japan制造的“irgacure oxe
‑
02”)、具有噻唑酮结构的2,4
‑
二甲基噻唑酮(日本化药株式会社制造的“detx
‑
s”)。此种利用光的自由基生成能力较高的光聚合引发剂在用于通常的丙烯酸化合物等的光聚合的情形下,存在反应性过高、反应控制较难的倾向,但在本发明中可适宜使用。
[0113]
<感光性树脂组成物>
[0114]
本发明的感光性树脂组成物含有上述双马来酰亚胺化合物(i)及上述光聚合引发剂(ii)。在本发明的感光性树脂组成物中,相对于100质量份的上述双马来酰亚胺化合物(i),上述光聚合引发剂(ii)的含量优选为0.1~15质量份,更优选为0.5~5质量份。在上述含量未达0.1质量份的情形下,存在于曝光时利用光照射的二聚反应未充分进行,从而在显影时聚合膜从无机表面保护膜剥离的倾向。另一方面,在上述含量超过15质量份的情形下,存在反应过度进行而进行未曝光部的聚合反应,因此难以形成微细图案的倾向。
[0115]
根据本发明的感光性树脂组成物,不需要热硬化,或者即便在视需要进行热硬化的情形下,也能够在与已知相比相对较低的温度下进行热硬化,可获得拉伸弹性模量充分小的硬化物。因此,可充分减小硬化后的膜内产生的残留应力,而可充分抑制硅晶圆等基板的翘曲。然后,根据本发明的感光性树脂组成物,即便为10μm以上的膜厚,也能够通过310~436nm(优选为365nm)的光照射形成微细的图案(优选为开口直径(via径)的纵横比为0.3以上,更优选为0.5以上)。此外,在本发明中,上述开口直径(via径)的纵横比是用下式:“纵横比=(硬化膜的膜厚)/(硬化膜中形成的贯通孔的开口直径)”表示的值。
[0116]
作为本发明的感光性树脂组成物,含有上述双马来酰亚胺化合物(i)、上述光聚合引发剂(ii)即可,并无特别限制,优选为利用有机溶剂使上述感光性树脂组成物溶解。作为上述有机溶剂,可列举:甲苯、二甲苯、四氢化萘等芳香族溶剂;甲基异丁基酮、环戊酮、环己酮等酮类溶剂;四羟基呋喃等环状醚类溶剂;苯甲酸甲酯等有机溶剂。作为这些有机溶剂,可单独使用一种,也可组合两种以上使用。然后,作为这些有机溶剂,也可在双马来酰亚胺化合物(i)不析出的范围内含有乳酸乙酯、丙二醇单甲醚乙酸酯、γ
‑
丁内酯等双马来酰亚胺化合物不易溶解的溶剂。作为使上述双马来酰亚胺化合物(i)、上述光聚合引发剂(ii)溶解于上述有机溶剂时的浓度,就成为适宜的粘度的观点而言,优选为以感光性树脂组成物的固形物成分浓度计为20~70质量%。
[0117]
另外,作为本发明的感光性树脂组成物,也可进一步含有增感剂。作为上述增感剂,可列举4,4
’‑
双(二乙氨基)二苯甲酮等。在本发明中,在含有上述增感剂的情形下,作为其含量,相对于100质量份的上述双马来酰亚胺化合物(i),优选为0.01~2质量份,更优选为0.05~0.5质量份。通过含有此种增感剂,可进一步提高感光性树脂组成物对光的感度。
[0118]
作为本发明的感光性树脂组成物,也可进一步含有聚合性化合物。上述聚合性化合物是指具有丙烯酰基、甲基丙烯酰基、烯丙基、苯乙烯基等聚合性官能团的化合物。上述聚合性化合物可为具有多种上述聚合性官能团的化合物。通过含有上述聚合性化合物,可进一步提高感光性树脂组成物对光的感度。作为上述聚合性化合物,就更容易引起利用光聚合的交联反应的观点而言,优选为丙烯酸酯。作为上述丙烯酸酯,可列举:二丙烯酸氢化二环戊二烯酯、丙烯酸二环戊烯酯、丙烯酸二环戊烯氧基乙酯、1,3
‑
丁二醇二丙烯酸酯、1,4
‑
丁二醇二丙烯酸酯、1,6
‑
丁二醇二丙烯酸酯、二乙二醇二丙烯酸酯、新戊二醇二丙烯酸酯、聚乙二醇200二丙烯酸酯、聚乙二醇400二丙烯酸酯、聚乙二醇600二丙烯酸酯、二乙二醇二丙烯酸酯、新戊二醇二丙烯酸酯、羟基新戊酸酯新戊二醇二丙烯酸酯、三乙二醇二丙烯酸酯、双(丙烯酰氧基乙氧基)双酚a、双(丙烯酰氧基乙氧基)四溴双酚a、三丙二醇二丙烯酸酯、三羟甲基丙烷三丙烯酸酯、季戊四醇三丙烯酸酯、异氰酸三(2
‑
羟基乙基)酯、季戊四醇四丙烯酸酯、二季戊四醇六丙烯酸酯、二季戊四醇单羟基五丙烯酸酯等。
[0119]
另外,在含有上述聚合性化合物的情形下,作为其含量,相对于100质量份的上述
双马来酰亚胺化合物(i),优选为30质量份以下。在上述聚合性化合物的含量超过30质量份的情形下,存在聚合性化合物单独进行利用光聚合的交联反应,所获得的硬化物的拉伸弹性模量变高的倾向。另外,因上述聚合性化合物对自由基反应性较高,故在使用如适宜用于本发明的具有选自由肟结构及噻唑酮结构所组成的群中的至少任一种结构的光聚合引发剂般反应性较高的光聚合引发剂的情形下,存在难以控制反应的倾向。此外,通常若添加聚合性化合物,则存在所获得的硬化物的拉伸弹性模量变高、柔软性受到损害的倾向,但本发明的双马来酰亚胺化合物(i)即便添加聚合性化合物,在所获得的硬化物中拉伸弹性模量也难以变高,柔软性难以受到损害。本发明人等推测其原因在于:本发明的双马来酰亚胺化合物(i)仅在两个末端具有反应性马来酰亚胺基团,而在分子链中不具有交联性反应基团。另外,作为本发明的感光性树脂组成物,在不阻碍本发明的效果的范围内,也可进一步含有调平剂、消泡剂等。
[0120]
本发明的感光性树脂组成物可通过通常已知的使用方法而使用。例如,首先,在将已利用上述有机溶剂调整粘度的本发明的感光性树脂组成物涂布在支持体后,在50~180℃、优选为80~140℃干燥5~30分钟,由此可制成膜状感光性树脂组成物。作为上述支持体,例如可列举:硅晶圆、陶瓷基板、刚性基板、可挠性基板及在硅晶圆上形成有sin膜或sio2膜等无机表面保护膜的支持体。根据本发明,即便使用上述形成有无机表面保护膜的硅晶圆作为支持体,也可获得与上述无机表面保护膜的密接性(接着性)优异的硬化物。
[0121]
作为上述涂布方法,并无特别限制,可列举使用旋转涂布机、狭缝式涂布机、辊式涂布机等的涂布、或网版印刷等。在这些方法中,例如作为对硅晶圆的涂布方法,优选采用使用旋转涂布机的涂布方法。另外,作为上述膜状感光性树脂组成物的膜厚,可通过调整上述感光性树脂组成物的浓度或涂布厚度而任意进行调整,并无特别限制,例如,在制成半导体组件用保护膜或层间绝缘膜的情形下,干燥后的膜厚优选为3~50μm,更优选为5~30μm,还更优选为5~20μm。在上述膜厚未达3μm的情形下,存在无法充分保护处于膜下的组件或电路的倾向,另一方面,在超过50μm的情形下,存在难以形成微细的图案的倾向。在本发明中,即便为10μm以上(优选为10~20μm)的膜厚,也能够形成微细的图案,且能够形成如通过下述曝光及显影形成的贯通孔的开口直径(via径)的纵横比为0.3以上(更优选为0.5以上)的图案。
[0122]
其次,对如此获得的膜状感光性树脂组成物适用具有特定图案形状的掩模进行曝光,使本发明的感光性树脂组成物进行光聚合。作为上述曝光方法,可列举接触曝光或缩小投影曝光。作为曝光波长,优选为200~500nm的紫外线至可见光,可使用标准使用的缩小投影曝光机(步进机)。另外,作为曝光波长,就可形成微细的图案的观点而言,更优选为310~436nm,还更优选为365nm。作为曝光量,并无特别限制,在本发明中,即便为相对较低的曝光量也能够形成微细的图案,无需大量曝光量,因此,优选为300~2000mj/cm2,更优选为500~1500mj/cm2。
[0123]
其次,进行利用显影液溶解去除上述曝光后的膜状感光性树脂组成物的未曝光部的显影,由此可获得具有特定图案的聚合膜(聚合物)。即,在曝光部中,因光照射而由光聚合引发剂产生的自由基与马来酰亚胺基团反应,主要通过二聚反应使上述双马来酰亚胺化合物(i)间交联,而对显影液不溶化。对此,由于未曝光部溶解在显影液,故而可通过利用曝光部与未曝光部对显影液的溶解度差而获得具有特定的开口直径(via径)的贯通孔等图案
的聚合膜。作为上述显影液,可列举:甲苯、二甲苯等芳香族类溶剂;环戊酮、环己酮等环状酮类溶剂;四羟基呋喃等环状醚类溶剂及它们的混合溶剂。另外,作为上述显影液,为了调整显影时的溶解性,也可进一步含有甲醇、乙醇、丙醇等醇类溶剂。作为上述显影方法,可列举喷雾法、覆液法、浸渍法等方法。
[0124]
另外,优选进一步利用环戊酮或环戊酮与乙醇的混合溶剂等有机溶剂对通过上述显影获得的具有特定图案的聚合膜进行冲洗。作为上述显影后的聚合膜,就抑制表面粗糙的产生,另外,尺寸设计变得容易的观点而言,优选残膜率为90%以上。在本发明中,上述残膜率是指显影后的聚合膜的膜厚相对于干燥后(曝光前)的膜状感光性树脂组成物的膜厚之比(显影后的聚合膜的膜厚/干燥后(曝光前)的膜状感光性树脂组成物的膜厚)。
[0125]
其次,可视需要对通过上述显影获得的具有特定图案的聚合膜进行加热而使其硬化,由此获得具有特定图案的硬化膜(硬化物)。作为上述加热温度(硬化温度),优选为60~230℃,更优选为150~230℃。另外,作为上述加热时间,优选为30~120分钟。此外,上述硬化温度是指本发明中通过热反应使在上述曝光时因未反应而残留的马来酰亚胺基团热硬化所需要的温度。通过此种热硬化反应使上述光聚合中未反应的马来酰亚胺基交联,但在使用本发明的感光性树脂组成物的情形下,无需如已知的聚酰亚胺前驱物或聚苯并噁唑前驱物般提高硬化温度。其原因在于:本发明的双马来酰亚胺化合物(i)中无需脱水闭环反应。
[0126]
如此,通过使用本发明的感光性树脂组成物,可获得具有微细图案的硬化膜。作为上述图案,所形成的贯通孔的开口直径(via径)的纵横比优选为0.3以上,更优选为0.5以上。此外,在本发明中,上述开口直径可通过使用光学显微镜或扫描式电子显微镜(sem)进行测定而求出。
[0127]
另外,在使用本发明的感光性树脂组成物获得的硬化膜中,拉伸弹性模量优选为50~800mpa,更优选为50~500mpa,还更优选为100~500mpa,还更优选为100~300mpa。如此使用本发明的感光性树脂组成物获得的硬化物由于上述硬化温度充分低,且拉伸弹性模量充分低,因此不会产生硅晶圆等基板的翘曲,其后的步骤中的操作性优异。此外,在本发明中,上述拉伸弹性模量可通过使用tensilon(拉伸试验机)在温度23℃、拉伸速度5mm/min的条件进行测定而求出。
[0128]
进而,在使用本发明的感光性树脂组成物获得的硬化膜中,就可抑制破裂的观点而言,断裂伸长率优选为20~200%,更优选为70%以上。此外,在本发明中,上述断裂伸长率可通过使用tensilon(拉伸试验机)在温度23℃、拉伸速度5mm/min的条件进行测定而求出。
[0129]
如上所述,根据本发明的感光性树脂组成物,能够实现与已知相比相对较低的温度下的热硬化及利用低曝光量的微细的图案形成,可获得拉伸弹性模量充分小、且与无机表面保护膜或金属配线材料的密接性优异的硬化物。另外,即便在热硬化的情形下,也能够在与已知相比相对较低的温度下进行热硬化,可获得拉伸弹性模量充分小的硬化膜,因此,可充分减小硬化后的膜内产生的残留应力,而可充分抑制硅晶圆等基板的翘曲。然后,根据本发明,即便在310~436nm(优选为365nm)的曝光波长、2000mj/cm2以下的低曝光量下也能够形成微细的图案,且能够形成如贯通孔的开口直径(via径)的纵横比为0.3以上(更优选为0.5以上)的图案。本发明人等推测其原因在于:本发明的感光性树脂组成物中365nm时的
吸收较少,且马来酰亚胺基团的反应主要为二聚反应,因此可抑制如丙烯酸化合物般因连锁反应而聚合进行至未曝光部。
[0130]
作为此种使用本发明的感光性树脂组成物获得的光硬化后或光热硬化(并用光硬化与热硬化的硬化)后的硬化物,可适宜用于选自由半导体组件的表面保护膜、层间绝缘膜及再配线层的绝缘膜所组成的群中的至少一种膜。另外,本发明的感光性树脂组成物在此种膜中必须为10μm以上的膜厚,且必须进行如贯通孔的开口直径(via径)的纵横比为0.3以上(更优选为0.5以上)的图案化的情形下尤其有效。
[0131]
以上,对于本发明的双马来酰亚胺化合物或感光性树脂组成物等进行了详述,关于利用本发明的感光性树脂组成物等达成本发明的目的的原因,本发明人等推测如下。即,已知的马来酰亚胺化合物通常在光聚合反应中主要进行二聚反应,因此与作为其它光聚合性化合物的丙烯酸化合物相比较,存在交联反应的效率较小的倾向。因此,本发明人等推测,为了充分形成利用光聚合的交联结构,需要非常多的曝光量。另外,已知马来酰亚胺化合物因化合物自身的光反应仅在310nm以下的波长下进行、以及难以引起利用自由基的连锁聚合等原因,而主要用作热聚合性化合物。与此相对,本发明的特定的双马来酰亚胺化合物为具有包含源自二聚酸的结构的柔软骨架的结构,因此通过将此种双马来酰亚胺化合物与例如产生自由基的光聚合引发剂组合,而马来酰亚胺基团彼此容易邻接,且交联反应的效率提高。因此推测,根据本发明的感光性树脂组成物,能够以相对较低的曝光量形成微细的图案,无需如已知的高温下的热硬化,进而可获得拉伸弹性模量充分小、且与被粘着体的密接性优异的硬化物。
[0132]
如此,本发明人等推测,利用本发明的感光性树脂组成物获得的硬化物的拉伸弹性模量充分小,由此可与被粘着体充分密接,由此也产生与无机表面保护膜或金属配线材料的相互作用,因此,与被粘着体、尤其是无机表面保护膜或金属配线材料的密接性优异。
[0133]
另外,本发明人等推测,本发明的感光性树脂组成物由于365nm时的吸收较少,故而即便膜厚为10μm以上,也能够使用半导体的保护膜等的制造步骤中标准地使用的缩小投影曝光机形成微细的图案。
[0134]
实施例
[0135]
以下,基于实施例及比较例更具体地说明本发明,但本发明并不限定于以下实施例。此外,各实施例及比较例中的图案化性能评估及机械物性评估分别以如下方式进行。
[0136]
分子量的测定条件如下所述。
[0137]
机种:gpc tosoh hlc
‑
8220gpc
[0138]
管柱:super hzm
‑
n
[0139]
溶离液:thf(四氢呋喃);0.35ml/min、40℃
[0140]
检测器:ri(示差折射计)
[0141]
分子量标准:聚苯乙烯
[0142]
合成例1(i
‑
1)
[0143]
向涂布有氟树脂的配备有搅拌棒的500ml圆底烧瓶中投入110g的甲苯与36g的n
‑
甲基吡咯烷酮。其次,加入88.0g(0.16mol)的priamine 1074(croda japan株式会社制造),然后,缓慢加入15.8g的(0.16mol)甲磺酸酐,以形成盐。大致搅拌10分钟加以混合,然后向经搅拌的混合物中缓慢加入5
‑
(2,5
‑
二氧四氢呋喃基)
‑3‑
甲基
‑3‑
环己烯
‑
1,2
‑
二羧酸二酐
(21.8g,0.08mol)。将迪安
‑
斯塔克分离器与冷凝器安装至烧瓶。将混合物加热6小时进行回流,形成胺封端的二酰亚胺。在此时之前获得来自该缩合的生成水的理论量。将反应混合物冷却至室温以下,向烧瓶中加入19.4g(0.20mol)的马来酸酐。将混合物进行回流8小时,获得期待量的生成水。冷却至室温后,然后向烧瓶中加入200ml的甲苯。然后,利用水(100ml
×
3次)清洗经稀释的有机层,去除盐或未反应的原料。其后,于真空下去除溶剂,获得琥珀色蜡状双马来酰亚胺化合物120g(产率95%,mw=3200)(i
‑
1)。
[0144]
合成例2(i
‑
2)
[0145]
向涂布有氟树脂的配备有搅拌棒的500ml圆底烧瓶中投入110g的甲苯与36g的n
‑
甲基吡咯烷酮。其次,加入90.5g(0.17mol)的priamine 1074(croda japan株式会社制造),然后,缓慢加入16.3g(0.17mol)的甲磺酸酐,以形成盐。大致搅拌10分钟加以混合,然后向经搅拌的混合物中缓慢加入1,2,4,5
‑
环己烷四羧酸二酐(18.9g,0.08mol)。将迪安
‑
斯塔克分离器与冷凝器安装至烧瓶。将混合物加热6小时进行回流,形成胺封端的二酰亚胺。在此时之前获得来自该缩合的生成水的理论量。将反应混合物冷却至室温以下,向烧瓶中加入19.9g(0.20mol)的马来酸酐。将混合物进行回流8小时,获得期待量的生成水。冷却至室温后,然后向烧瓶中加入200ml的甲苯。然后,利用水(100ml
×
3次)清洗经稀释的有机层,去除盐或未反应的原料。其后,在真空下去除溶剂,获得琥珀色蜡状双马来酰亚胺化合物110g(产率92%,mw=3000)
ꢀꢀꢀꢀ
(i
‑
2)。
[0146]
合成例3(i
‑
3)
[0147]
向涂布有氟树脂的配备有搅拌棒的500ml圆底烧瓶中投入110g的甲苯与36g的n
‑
甲基吡咯烷酮。其次,加入85.6g(0.16mol)的priamine 1074(croda japan株式会社制造),然后,缓慢加入15.4g(0.16mol)的甲磺酸酐,以形成盐。大致搅拌10分钟加以混合,然后向经搅拌的混合物中缓慢加入1,1
’‑
双环己烷
‑
3,3’,4,4
’‑
四羧酸
‑
3,4:3’,4
’‑
二酐(24.5g,0.08mol)。将迪安
‑
斯塔克分离器与冷凝器安装至烧瓶。将混合物加热6小时进行回流,形成胺封端的二酰亚胺。在此时之前获得来自该缩合的生成水的理论量。将反应混合物冷却至室温以下,向烧瓶中加入18.8g(0.19mol)的马来酸酐。将混合物进行回流8小时,获得期待量的生成水。冷却至室温后,然后向烧瓶中加入200ml的甲苯。然后,利用水(100ml
×
3次)清洗经稀释的有机层,去除盐或未反应的原料。其后,在真空下去除溶剂,获得琥珀色蜡状双马来酰亚胺化合物108g(产率90%,mw=3600)
ꢀꢀꢀꢀ
(i
‑
3)。
[0148]
合成例4(i
‑
4)
[0149]
向涂布有氟树脂的配备有搅拌棒的500ml圆底烧瓶中投入110g的甲苯与36g的n
‑
甲基吡咯烷酮。其次,加入85.9g(0.16mol)的priamine 1074(croda japan株式会社制造),然后,缓慢加入15.5g(0.16mol)的甲磺酸酐,以形成盐。大致搅拌10分钟加以混合,然后向经搅拌的混合物中缓慢加入4
‑
(2,5
‑
二氧四氢呋喃
‑3‑
基)
‑
1,2,3,4
‑
四氢化萘
‑
1,2
‑
二羧酸酐(24.1g,0.08mol)。将迪安
‑
斯塔克分离器与冷凝器安装至烧瓶。将混合物加热6小时进行回流,形成胺封端的二酰亚胺。在此时之前获得来自该缩合的生成水的理论量。将反应混合物冷却至室温以下,向烧瓶中加入18.9g(0.19mol)的马来酸酐。将混合物进行回流8小时,获得期待量的生成水。冷却至室温后,然后向烧瓶中加入200ml的甲苯。然后,利用水(100ml
×
3次)清洗经稀释的有机层,去除盐或未反应的原料。其后,在真空下去除溶剂,获得暗琥珀色蜡状双马来酰亚胺化合物106g(产率89%,mw=3700)(i
‑
4)。
[0150]
合成例5(i
‑
5)
[0151]
向涂布有氟树脂的配备有搅拌棒的500ml圆底烧瓶中投入110g的甲苯与36g的n
‑
甲基吡咯烷酮。其次,加入73.5g(0.14mol)的priamine 1074(croda japan株式会社制造)与8.4g(0.06mol)的1,3
‑
双(氨基甲基)环己烷,然后,缓慢加入18.9g(0.20mol)的甲磺酸酐,以形成盐。大致搅拌10分钟加以混合,然后向经搅拌的混合物中缓慢加入5
‑
(2,5
‑
二氧四氢呋喃基)
‑3‑
甲基
‑3‑
环己烯
‑
1,2
‑
二羧酸二酐(26.0g,0.10mol)。将迪安
‑
斯塔克分离器与冷凝器安装至烧瓶。将混合物加热6小时进行回流,形成胺封端的二酰亚胺。在此时之前获得来自该缩合的生成水的理论量。将反应混合物冷却至室温以下,向烧瓶中加入23.1g(0.24mol)的马来酸酐。将混合物进而回流8小时,获得期待量的生成水。冷却至室温后,然后向烧瓶中加入200ml的甲苯。然后,利用水(100ml
×
3次)清洗经稀释的有机层,去除盐或未反应的原料。其后,在真空下去除溶剂,获得琥珀色蜡状双马来酰亚胺化合物108g(产率90%,mw=2800)(i
‑
5)。
[0152]
比较合成例1
[0153]
向涂布有氟树脂的配备有搅拌棒的500ml圆底烧瓶中投入110g的甲苯与36g的n
‑
甲基吡咯烷酮。其次,加入90.9g(0.17mol)的priamine 1074(croda japan株式会社制造),然后,缓慢加入16.4g(0.17mol)的甲磺酸酐,以形成盐。大致搅拌10分钟加以混合,然后向经搅拌的混合物中缓慢加入均苯四甲酸酐(18.6g,0.08mol)。将迪安
‑
斯塔克分离器与冷凝器安装至烧瓶。将混合物加热6小时进行回流,形成胺封端的二酰亚胺。在此时之前获得来自该缩合的生成水的理论量。将反应混合物冷却至室温以下,向烧瓶中加入20.0g(0.20mol)的马来酸酐。将混合物进行回流8小时,获得期待量的生成水。冷却至室温后,然后向烧瓶中加入200ml的甲苯。然后,利用水(100ml
×
3次)清洗经稀释的有机层,去除盐或未反应的原料。其后,在真空下去除溶剂,获得褐色蜡状双马来酰亚胺化合物102g(产率85%,mw=3800)。
[0154]
比较合成例1的双马来酰亚胺化合物可作为“bmi
‑
3000”而容易地从designer molecures inc.公司获取。
[0155]
比较合成例2
[0156]
向涂布有氟树脂的配备有搅拌棒的500ml圆底烧瓶中投入110g的甲苯与36g的n
‑
甲基吡咯烷酮。其次,加入85.3g(0.16mol)的priamine 1074(croda japan株式会社制造),然后,缓慢加入15.4g(0.16mol)的甲磺酸酐,以形成盐。大致搅拌10分钟加以混合,然后向经搅拌的混合物中缓慢加入4,4
’‑
氧双邻苯二甲酸二酐(24.8g,0.08mol)。将迪安
‑
斯塔克分离器与冷凝器安装至烧瓶。将混合物加热6小时进行回流,形成胺封端的二酰亚胺。在此时之前获得来自该缩合的生成水的理论量。将反应混合物冷却至室温以下,向烧瓶中加入18.8g(0.19mol)的马来酸酐。将混合物进行回流8小时,获得期待量的生成水。冷却至室温后,然后向烧瓶中加入200ml的甲苯。然后,利用水(100ml
×
3次)清洗经稀释的有机层,去除盐或未反应的原料。其后,在真空下去除溶剂,获得褐色蜡状双马来酰亚胺化合物106g(产率88%,mw=3700)。
[0157]
比较合成例2的双马来酰亚胺化合物可作为“bmi
‑
1500”而容易地从designer molecures inc.公司获取。
[0158]
比较合成例3
[0159]
向涂布有氟树脂的配备有搅拌棒的500ml圆底烧瓶中投入110g的甲苯与36g的n
‑
甲基吡咯烷酮。其次,加入90.9g(0.17mol)的priamine 1074(croda japan株式会社制造),然后,缓慢加入16.4g(0.17mol)的甲磺酸酐,以形成盐。大致搅拌10分钟加以混合,继而向经搅拌的混合物中缓慢加入均苯四甲酸酐(18.6g,0.08mol)。将迪安
‑
斯塔克分离器与冷凝器安装至烧瓶。将混合物加热6小时进行回流,形成胺封端的二酰亚胺。在此时之前获得来自该缩合的生成水的理论量。冷却至室温后,然后向烧瓶中加入200ml的甲苯。然后,利用水(100ml
×
3次)清洗经稀释的有机层,去除盐或未反应的原料。其后,在真空下去除溶剂,获得褐色蜡状聚酰亚胺化合物90.4g(产率85%,mw=3600)。
[0160]
示出本实施例中使用的材料。
[0161]
[(i)成分:双马来酰亚胺化合物]
[0162]
i:合成例(i
‑
1)~(i
‑
5)所表示的双马来酰亚胺化合物及比较合成例1~3所表示的双马来酰亚胺化合物、聚酰亚胺化合物
[0163]
[(ii)成分:光聚合引发剂]
[0164]
ii
‑
1:乙酮,1
‑
[9
‑
乙基
‑6‑
(2
‑
甲基苯甲酰基)
‑
9h
‑
咔唑
‑3‑
基]
‑
,1
‑
(o
‑
乙酰肟)(basf japan制造的“irgacure oxe
‑
02”)
[0165]
ii
‑
2:2,4
‑
二甲基噻唑酮(日本化药株式会社制造的“detx
‑
s”)
[0166]
(实施例1~5及比较例1~3)
[0167]
调配表1中所示的调配量(质量份)的(i)~(ii)成分、50质量份的作为溶剂的环戊酮,制备实施例1~5及比较例1~3的感光性树脂组成物。
[0168]
<感光性树脂组成物的评估>
[0169]
对于实施例1~5及比较例1~3的感光性树脂组成物,进行如下所示的评估。将其结果汇总示于表1。
[0170]
[表1]
[0171][0172]
*1:3000mj/cm2时无法获得硬化膜。
[0173]
*2:无法获得硬化膜,因此未测定。
[0174]
(感度、残膜率、分辨率、显影残渣)
[0175]
将实施例1~5及比较例1~3中获得的感光性树脂组成物旋转涂布于硅基板上,在120℃加热4分钟,形成膜厚为10~15μm的涂膜。其次,使用ushio制造的“超高压水银灯500w multi
‑
light”,利用i射线(365nm)经由具有长为1μm、宽为1μm至长为100μm、宽为100μm的正方形孔图案的掩模进行缩小投影曝光。一边以每次改变100mj/cm2的方式将曝光量自500mj/cm2至3000mj/cm2改变一边进行曝光。曝光后,使用环戊酮进行显影。感度设为残膜率开始成为一定的曝光量。此外,残膜率通过下式算出。
[0176]
残膜率(%)=(显影后的涂膜的膜厚/显影前的涂膜的膜厚)
×
100
[0177]
表1的残膜率为表1中记载的感度的残膜率。
[0178]
另外,将开口的正方形孔图案中的最小开口宽度设为分辨率的指标。此外,感度及分辨率越小越佳。将结果示于表1。
[0179]
然后,在利用显微镜观察显影后的图案时,将在图案开口部的整体或者一部分中发现残渣的在显影残渣的项目中评估为
×
。将无残渣的设为
○
。
[0180]
其后,将抗蚀图案在氮气中、温度180℃加热处理(硬化)60分钟。
[0181]
(机械物性评估)
[0182]
首先,在厚度为12μm的铜箔上使用旋转涂布机涂布各实施例及比较例中获得的感光性树脂组成物后,在温度100℃干燥10分钟,在铜箔上形成膜状感光性树脂组成物。以干燥后的膜状感光性树脂组成物的膜厚成为10μm的方式调整感光性树脂组成物的涂布厚度。对于该膜状感光性树脂组成物,使用ushio制造的“超高压水银灯500w multi
‑
light”,在波长365nm、曝光量2000mj/cm2下进行曝光,其次,在温度180℃加热60分钟进行硬化后,通过蚀刻去除铜箔,由此获得硬化膜。
[0183]
然后,将所获得的硬化膜切割成10mm的长度,在温度23℃下使用tensilon(拉伸试验机)在拉伸速度为5mm/min的条件测定断裂伸长率(%)及拉伸弹性模量(mpa)而求出。
[0184]
(介电特性(介电常数:dk、介电损耗正切:df)评估)
[0185]
为了评估介电特性,通过桌上型涂布机以干燥后的厚度为50μm的方式将清漆涂敷于铜箔上并加以干燥,获得树脂膜(半硬化)。然后,对所获得的树脂膜(半硬化)照射2000mj/cm2的uv。在所制造的树脂膜上同样地形成树脂膜并积层,将树脂膜的膜厚设为300μm。然后,通过物理剥离或者蚀刻去除作为支持体的铜箔,从而获得评估用树脂膜。
[0186]
然后,将树脂膜切割为长60mm、宽2mm、厚0.3mm,以所获得的作为试片,通过空腔共振器扰动法测定介电特性。测定器使用aet公司制造的向量型网络分析仪admso10c1,空腔共振器使用关东电子应用开发株式会社制造的cp531(10ghz带共振器)。条件设为频率10ghz、测定温度25℃。
[0187]
(吸水率的测定)
[0188]
使用棒式编码器,在无锡钢板上以200μm的厚度涂布清漆,在90℃干燥5分钟,形成树脂层。以2000mj/cm2进行曝光而硬化后,在180℃加热1小时,由此制作试样(硬化物)。将该硬化膜在25℃的水中浸渍24小时,从水中取出,充分擦去水,利用卡氏法计算硬化膜中的水分量。
[0189]
(hast耐性)
[0190]
通过网版印刷法以成为25微米的厚度的方式将各组成物涂布在形成有l/s=10μm/10μm的梳型图案的espanex m series(新日铁化学制造:基底酰亚胺厚25μm,cu厚18μm)上,利用80℃的热风干燥器将涂膜干燥60分钟。其次,使用紫外线曝光装置(ushio制造:500w multi
‑
light)以2000mj/cm2进行曝光而硬化后,在180℃加热1小时,由此获得hast评估用试验基板。对获得的基板的电极部分进行利用焊料的配线连接,置于130℃、85%rh的环境,施加5.5v的电压,测定直至电阻值成为1
×
108ω以下的时间。
[0191]
○
:300小时以上
[0192]
δ:30~300小时
[0193]
×
:30小时以下
[0194]
根据表1所示的结果可知,在使用实施例1~5中获得的本发明的感光性树脂组成物的情形下,确认到即便在低曝光量下,也能够获得充分小的开口直径,从而形成微细的图案。另外,确认到实施例1~5中获得的本发明的感光性树脂组成物即便不进行利用高温热硬化,也可获得拉伸弹性模量充分小且与无机表面保护膜等被粘着体的密接性优异的硬化物。与此相对,在使用比较例1~3中获得的感光性树脂组成物的情形下,确认到形成图案需要高达3000mj/cm2的曝光量,难以用作感光性树脂组成物。
[0195]
另外,表1所示的结果清楚地表明,使用本发明的感光性树脂组成物获得的硬化物为如下优异的马来酰亚胺化合物,其即便在低曝光量下也充分进行马来酰亚胺基团的光硬化,由此可保持较低的介电特性及吸水率,并且保持较高的绝缘可靠性。
[0196]
本技术基于2019年4月2日提出申请的日本专利申请(日本特愿2019
‑
070316号),将其内容作为参照并入本文。
[0197]
产业上的实用性
[0198]
如以上所说明,根据本发明,能够提供一种感光性树脂组成物、使用其的硬化物及半导体组件;上述感光性树脂组成物能够在相对较低的曝光量(2000mj/cm2以下)形成微细的图案,且无需进行如已知的高温下的热硬化,而可获得拉伸弹性模量充分小、与无机表面保护膜(氮化硅膜、氧化硅膜等)或导电性金属配线材料(铜等)的密接性优异的硬化物。
[0199]
另外,根据本发明,无需热硬化,即便在视需要进行热硬化的情形下,也能够在与已知相比相对较低的温度(60~230℃)进行热硬化,从而可获得拉伸弹性模量充分小的硬化物,因此,可充分减小硬化后的膜内产生的残留应力,从而可充分抑制硅晶圆的翘曲。然后,根据本发明,即便膜厚较厚,也能够通过365nm的光照射形成微细的图案。因此,此种本发明的感光性树脂组成物作为半导体组件的表面保护膜、层间绝缘膜及再配线层的绝缘膜等非常有用。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。