1.本发明涉及一种药材中有关微量物质的检测方法,特别涉及一种检测中药材银杏叶中咖 啡因含量的方法。
背景技术:
2.银杏叶,为银杏科植物银杏ginkgo biloba l.的干燥叶。秋季叶尚绿时采收,及时干燥。 具有活血化瘀,通络止痛,敛肺平喘,化浊降脂的功效。用于瘀血阻络,胸痹心痛,中风偏 瘫,肺虚咳喘,高脂血症。
3.2015年版《中国药典》中写明,银杏叶中含总黄酮醇苷类、萜类内酯,后者主要包括银 杏内酯a(c
20
h
24
o9)、银杏内酯b(c
20
h
24
o
10
)、银杏内酯c(c
20
h
24
o
11
)和白果内酯(c
15
h
18
o8)。
4.现代药理研究表明,银杏叶具有抗氧化、抗凋亡、改善脑血流、神经保护、抑制血小板 活性等多种药理作用,可用于心脑血管等疾病的治疗。
5.因此,银杏叶及银杏叶制剂被广泛应用于临床。
6.近年来,有研究先后运用高效液相色谱-串联质谱法和超高效液相-串联质谱法分别检测 出银杏叶制剂、银杏叶中含有咖啡因成分,咖啡因作为第二类精神药品,其影响包括心悸、 心率失常、中枢神经系统过度兴奋等不良反应,尤其是其具有成瘾性,可能对人体造成危害。
7.由于银杏叶中咖啡因含量极少,因此建立一种简单快速的检测方法专门用来测定银杏叶 中咖啡因的含量十分必要。
8.咖啡因的检测方法已有报道,如:食品中西布曲明等化合物的测定(bjs 201701),该方 法用来规定并检测食品(含保健食品)基质中西布曲明等(含咖啡因)33种违法添加化合物 的高效液相色谱-串联质谱测定方法,该方法得到的图谱、数据质量较好,结果准确、可靠, 并且其定量的线性范围下限达到0.1ng/ml。但是,该方法所检测的咖啡因是外源性物质,无 法直接用于银杏叶。
技术实现要素:
9.本发明提供了一种银杏叶药材中微量成分咖啡因含量的测定方法,所述方法以bjs201701方法(食品中西布曲明等化合物的测定(bjs 201701,简称文献1)为基础,对其检 测条件进行了改进,以适应银杏叶药材,同时,筛选得到特别优选的仪器,溶剂,流动相, 温度,时间等条件,特别适合银杏叶药材的检测。
10.本发明的检测方法,采用超高效液相色谱-串联质谱测定方法,其中,采用特定仪器, waters acquity uplc i-class超高效液相色谱仪串联xevo tq-s micro三重四极杆质谱仪,进 行测定。
11.本发明所述的方法,包括以下步骤:
12.1)供试品溶液的制备:
13.2)对照品溶液的制备:
14.3)检测:取供试品溶液和对照品溶液,注入液质联用仪,以咖啡因对照品峰为参照峰, 记录峰面积,根据峰面积计算供试品溶液中的咖啡因含量。
15.优选的,本发明所述的方法,包括以下步骤:
16.1)供试品溶液的制备:取银杏叶样品加入甲醇提取,提取液过滤,取续滤液离心得到上 清液;
17.2)对照品溶液的制备:取咖啡因对照品用甲醇溶解并稀释至刻度;
18.3)检测取供试品溶液和对照品溶液,注入液质联用仪,以咖啡因对照品峰为参照峰,记 录峰面积,根据峰面积计算供试品溶液中的咖啡因含量,其中,液质联用仪的色谱、质谱条 件采用的仪器是waters acquity uplc i-class超高效液相色谱仪串联xevo tq-s micro三重四 极杆质谱仪,
19.其中,所述液质联用仪的液相色谱条件如下:色谱柱:waters cortecs t3色谱柱(2.1
×
100 mm,2.7μm);流动相:a为含0.1%甲酸水溶液,b为含0.1%甲酸乙腈溶液,梯度洗脱,0~5 min,95%a;5~22min,95%~2%a;22~27min,2%a;27~27.5min,2%~95%a; 27.5~32min,95%a;流速:0.3ml/min;柱温30℃;进样量2μl;质谱参数条件:正离 子模式;毛细管电压:0.5kv;离子源温度:150℃;雾化温度:500℃;雾化器流速:1000 l/h;多反应监测模式(mrm);离子对195.112
→
138.065(定量)、195.112
→
110.037(定性); 驻留时间分别为0.165s、0.165s;锥孔电压分别为4.0v、4.0v;碰撞能量分别为18.0v、22.0 v。
20.具体的,本发明所述的方法,其中,
21.步骤1)供试品溶液的制备方法如下:取银杏叶样品适量,研成粉末,过四号筛,混匀, 称取粉末1.0g,精密称定,置具塞锥形瓶中,精密加入甲醇8-12ml,密塞,称重,超声提 取8-12min,放冷,再次称重,用甲醇补足减失的重量,摇匀,用0.2μm针筒式过滤器过 滤,取续滤液,高速离心10-20min,转数10000-15000转/分钟,得上清液,即为供试品溶液。
22.其中,步骤2)对照品溶液的制备方法如下:取咖啡因对照品2.50mg,精密称定,置于 5ml容量瓶中,用甲醇溶解并稀释至刻度,摇匀,制成浓度为500μg/ml咖啡因对照品溶 液。
23.更具体的,本发明所述的方法,其中1)供试品溶液的制备方法如下:取银杏叶样品适 量,研成粉末,过四号筛,混匀,称取粉末1.0g,精密称定,置具塞锥形瓶中,精密加入甲醇 10ml,密塞,称重,超声提取10min,放冷,再次称重,用甲醇补足减失的重量,摇匀, 用0.2μm针筒式过滤器过滤,取续滤液,高速离心15min,转数12000转/分钟,得上清液 即为供试品溶液。
24.其中,步骤2)对照品溶液的制备方法如下:取咖啡因对照品2.50mg,精密称定,置于 5ml容量瓶中,用甲醇溶解并稀释至刻度,摇匀,制成浓度为500μg/ml咖啡因对照品溶 液。
25.其中步骤3)色谱、质谱条件采用的仪器是waters acquity uplc i-class超高效液相色谱 仪串联xevo tq-s micro三重四极杆质谱仪,具体参数如下:
26.液相色谱条件:色谱柱:waters cortecs t3色谱柱(2.1
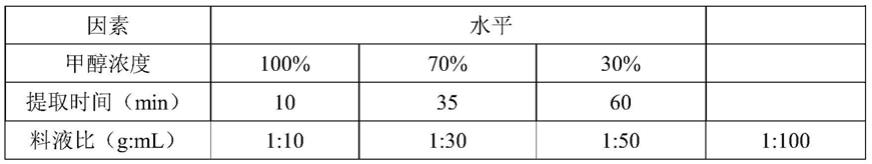
×
100mm,2.7μm);流动相:a 为含0.1%甲酸水溶液,b为含0.1%甲酸乙腈溶液,梯度洗脱,0~5min,95%a;5~22min, 95%~2%a;22~27min,2%a;27~27.5min,2%~95%a;27.5~32min,95%a;流 速:0.3ml/min;柱温30℃;进样量2μl
27.质谱参数条件:正离子模式;毛细管电压:0.5kv;离子源温度:150℃;雾化温度: 500℃;雾化器流速:1000l/h;多反应监测模式(mrm);离子对195.112
→
138.065(定量)、 195.112
→
110.037(定性);驻留时间分别为0.165s、0.165s;锥孔电压分别为4.0v、4.0v; 碰撞能量分别为18.0v、22.0v
28.特别优选的,本发明所述的方法,包括以下步骤:
29.1)供试品溶液的制备方法如下:取银杏叶样品适量,研成粉末,过四号筛,混匀,称取 粉末1.0g,精密称定,置具塞锥形瓶中,精密加入甲醇8-12ml,密塞,称重,超声提取8-12 min,放冷,再次称重,用甲醇补足减失的重量,摇匀,用0.2μm针筒式过滤器过滤,取续 滤液,高速离心10-20min,转数10000-15000转/分钟,得上清液,即为供试品溶液;
30.2)对照品溶液的制备方法如下:取咖啡因对照品2.50mg,精密称定,置于5ml容量 瓶中,用甲醇溶解并稀释至刻度,摇匀,制成浓度为500μg/ml咖啡因对照品溶液;
31.3)取供试品溶液和对照品溶液,注入液质联用仪,以咖啡因对照品峰为参照峰,记录峰 面积,根据峰面积计算供试品溶液中的咖啡因含量;
32.其中,液相色谱条件:色谱柱:waters cortecs t3色谱柱(2.1
×
100mm,2.7μm);流动 相:a为含0.1%甲酸水溶液,b为含0.1%甲酸乙腈溶液,梯度洗脱,0~5min,95%a;5~22 min,95%~2%a;22~27min,2%a;27~27.5min,2%~95%a;27.5~32min,95%a; 流速:0.3ml/min;柱温30℃;进样量2μl;质谱参数条件:正离子模式;毛细管电压:0.5 kv;离子源温度:150℃;雾化温度:500℃;雾化器流速:1000l/h;多反应监测模式(mrm); 离子对195.112
→
138.065(定量)、195.112
→
110.037(定性);驻留时间分别为0.165s、0.165 s;锥孔电压分别为4.0v、4.0v;碰撞能量分别为18.0v、22.0v。
33.最优选的,本发明所述的方法,包括以下步骤:
34.1)供试品溶液的制备方法如下:取银杏叶样品适量,研成粉末,过四号筛,混匀,称取 粉末1.0g,精密称定,置具塞锥形瓶中,精密加入甲醇10ml,密塞,称重,超声提取10min, 放冷,再次称重,用甲醇补足减失的重量,摇匀,用0.2μm针筒式过滤器过滤,取续滤液, 高速离心15min,转数12000转/分钟,得上清液即为供试品溶液;
35.2)对照品溶液的制备方法如下:取咖啡因对照品2.50mg,精密称定,置于5ml容量 瓶中,用甲醇溶解并稀释至刻度,摇匀,制成浓度为500μg/ml咖啡因对照品溶液;
36.3)取供试品溶液和对照品溶液,注入液质联用仪,以咖啡因对照品峰为参照峰,记录峰 面积,根据峰面积计算供试品溶液中的咖啡因含量;
37.其中3)色谱、质谱条件采用的仪器是waters acquity uplc i-class超高效液相色谱仪串 联xevo tq-s micro三重四极杆质谱仪,具体参数如下:
38.液相色谱条件:色谱柱:waters cortecs t3色谱柱(2.1
×
100mm,2.7μm);流动相:a 为含0.1%甲酸水溶液,b为含0.1%甲酸乙腈溶液,梯度洗脱,0~5min,95%a;5~22min, 95%~2%a;22~27min,2%a;27~27.5min,2%~95%a;27.5~32min,95%a;流 速:0.3ml/min;柱温30℃;进样量2μl
39.质谱参数条件:正离子模式;毛细管电压:0.5kv;离子源温度:150℃;雾化温度: 500℃;雾化器流速:1000l/h;多反应监测模式(mrm);离子对195.112
→
138.065(定量)、 195.112
→
110.037(定性);驻留时间分别为0.165s、0.165s;锥孔电压分别为4.0v、4.0v; 碰撞能量分别为18.0v、22.0v。
40.本发明的的检测方法,是经过筛选获得的,筛选过程如下:
41.一、现有方法的改进
42.bjs 201701方法(食品中西布曲明等化合物的测定(bjs 201701,简称文献1)检测的 对象为食品(含保健食品)这一类物质,在对银杏叶中的咖啡因进行检测时,直接用文献1 方法检测结果较差,具体原因前面已经论述,今,发明人对bjs 201701方法进行改进,方法 检测的对象针对银杏叶生药样品。主要有:
43.1、进样量的改进
44.采用文献1中供试品溶液的制备、对照品溶液的制备、色谱质谱条件。
45.对进样量进行筛选,结果显示:0.1ng/ml咖啡因对照品进样量为1μl时峰面积为361, 信噪比为12.21,仅略超过定量限s/n=10,由于信噪比的计算结果具有一定的波动幅度,导 致结果不准;当进样量为2μl时峰面积为618,信噪比为20.24,超过定量限2倍,用于定量 结果较为准确。进样量大于等于2μl,均能满足实验要求,且2μl更节省样品用量,对仪器 和色谱柱的污染程度更小。
46.2、提取容器的改变
47.文献1的容器为具塞试管,由于具塞试管形状狭长,银杏叶药材粉末堆积在试管底部, 超声提取时,粉末多在下部,溶剂多在上部,使得每批银杏叶粉末与溶剂接触充分程度不一 致,导致结果差异性较大。而用具塞锥形瓶,超声时药材粉末与提取溶剂接触充分,结果稳 定性较好。
48.用具塞试管为提取容器的数据(同一批银杏叶样品平行实验结果)结果分别为255、372、 2443。峰面积波动很大,rsd值为123.28%。
49.用具塞锥形瓶为提取容器的数据(同一批银杏叶样品平行实验结果)结果分别为3029、 3057、3003。峰面积rsd值为0.89%。
50.3、样品处理的改进
51.文献1中将食品(含保健食品)直接研细使用,无去除杂质、过筛等步骤。由于银杏叶 药材含有杂质,使用文献1中方法会因杂质的存在干扰检测结果,使检测结果不准确。本方 法中取银杏叶药材,除去非银杏叶药用部分的杂质后,取全叶用打粉机粉碎,过四号筛,备 用。
52.文献1方法中使用“用微孔滤膜过滤,取续滤液,根据实际浓度适当稀释至线性范围内, 备用”,本发明方法中“用0.2μm针筒式过滤器过滤,取续滤液,高速离心15min,转数 12000转/分钟,取上清液,备用”,本方法增加离心、取上清液步骤,进一步去除银杏叶样 品中的杂质,使检测结果更准确,同时也减少样品对色谱柱、仪器的污染程度。
53.二、银杏叶提取工艺条件的确定
54.以甲醇浓度、提取时间、料液比为因素条件进行单因素试验,分别考察各因素对银杏叶 中咖啡因提取率的影响,见表1。
55.表1:单因素试验因素和水平
[0056][0057]
1、甲醇浓度对银杏叶中咖啡因提取率的影响
[0058]
称取1g银杏叶粉末,提取时间为10min,料液比(g:ml)为1:10,超声温度为30℃时, 采用100%、70%、30%体积分数的甲醇溶液提取银杏叶中咖啡因,以甲醇浓度为横坐标,咖 啡因提取率(ng/g)为纵坐标作图,见图1-1。
[0059]
由图1-1可知,甲醇浓度为100%、70%时提取率较高,提取时选择二者皆可,又考虑到 溶剂回收的方便性,选用100%甲醇则更有利于甲醇溶剂的回收,因此本实验选用100%甲醇 作为提取溶剂。
[0060]
2、不同提取时间对银杏叶中咖啡因提取率的影响
[0061]
称取1g银杏叶粉末,甲醇浓度为100%,料液比(g:ml)为1:10,超声温度为30℃时, 分别超声10min、35min、60min提取银杏叶中咖啡因,以提取时间为横坐标,咖啡因提取 率(ng/g)为纵坐标作图,见图1-2。
[0062]
由图1-2可知,提取时间为10min时提取率最高,且超声10min具有节约时间、成本的 优点,因此本实验选择提取时间为超声10min。
[0063]
3、不同料液比对银杏叶中咖啡因提取率的影响
[0064]
称取1g银杏叶粉末,甲醇浓度为100%,提取时间为10min,超声温度为30℃时,分 别采用料液比(g:ml)为1:10、1:30、1:50、1:100的溶剂量提取银杏叶中咖啡因,以料液比 (g:ml)为横坐标,咖啡因提取率(ng/g)为纵坐标作图,见图1-3。
[0065]
由图1-3可知,料液比在1:10~1:100的范围内,随着料液比的增大,提取率越高,且提 取率均较好,由此可知料液比在1:10~1:100的范围内皆可,在料液比为1:10时,峰面积的响 应值最高,且更节省溶剂,因此本实验选用的料液比为1:10(即称1g样品,溶剂用量为10ml)。
[0066]
综上,因此本实验采用100%甲醇超声提取10min,料液比为1:10。
[0067]
三、现有方法改进二
[0068]
本发明的方法与现有文献“超高效液相-串联质谱法研究银杏叶提取物提取过程咖啡因的 转移规律(张苹苹等,食品工业科技,2019年22期,34-49,下面简称文献2)”方法的比较
[0069]
1、提取溶剂单一,溶剂用量少,提取时间短,提取方法简单。
[0070]
(1)现有文献2中银杏叶的提取方法:准确称取1.0g(精确到0.0001)银杏叶,置于 250ml锥形瓶中,加入100ml 30%甲醇溶液超声1h,超声温度为30℃
±
0.5℃,根据实 际浓度适当稀释至线性范围内,用0.22μm滤膜过滤,装小瓶,供高效液相色谱-串联质谱 仪测定。
[0071]
(2)本发明方法:取银杏叶样品适量,研成粉末,过四号筛,混匀。称取粉末1.0g, 精密称定,置具塞锥形瓶中,精密加入甲醇10ml,密塞,称重,超声提取10min,放冷, 再次称重,用甲醇补足减失的重量,摇匀,用0.2μm针筒式过滤器过滤,取续滤液,高速离 心15min,
转数12000转/分钟,取上清液,备用。
[0072]
(3)对比:
[0073]
a文献2需甲醇30ml(提取溶剂为30%甲醇溶液,100ml溶液中含有30ml甲醇), 本发明方法仅需甲醇10ml,节省了2倍的甲醇用量。
[0074]
b文献2提取的溶剂为30%甲醇水溶液,本发明方法提取溶剂为纯甲醇。
[0075]
c同样为超声提取,文献2提取时间为1h,而本发明方法仅需超声10min,节约时间50 min。
[0076]
优势:本方法与现有文献2的方法在都能检测出银杏叶中咖啡因的基础上,具有节省溶 剂、溶剂单一、节省时间、回收便捷的优点,可用于快速、简便地检测银杏叶中的咖啡因等 成分。
[0077]
四、总结
[0078]
1、用本方法检测银杏叶中的咖啡因,检测限、定量限均低于现有方法。
[0079]
(1)现有文献2方法检测限为5μg/kg,定量限为15μg/kg。
[0080]
(2)本发明方法分别以信噪比s/n=3、s/n=10定义检测限和定量限,检测限、定量限 均在0.1ng/ml(即1μg/kg)以下。
[0081]
2、本发明方法得到的咖啡因色谱峰峰形较好,目标峰与杂质峰分离效果明显。
[0082]
采用文献2的方法得到的图见图2-1
[0083]
本发明方法得到的图,见图2-2、2-3、2-4分别为甲醇空白溶剂、咖啡因对照品溶液、银 杏叶供试品溶液在测定条件下的多反应监测模式色谱图。该图表明在选用离子对 195.11
→
138.065(定量)、195.112
→
110.037(定性)的测定条件下溶剂中无杂峰影响咖啡因 的测定,咖啡因对照品溶液中咖啡因的色谱峰不受杂质干扰、峰形优美,银杏叶供试品溶液 中咖啡因对应的峰与对照品峰保留时间一致,峰形也一致。该图充分说明该方法的有效、可 靠与成熟性。
[0084]
本发明提供的方法具有以下优点:
[0085]
1、方法比较
[0086]
本发明提供的方法较为成熟,是以《食品中西布曲明等化合物的测定(bjs 201701)》这 一标准方法为基础来建立的方法,相对来说,银杏叶中的咖啡因的检测比较难,原因:1)含 量极少(约1.5260
×
10-6
g/kg~61.1070
×
10-6
g/kg);2)银杏叶粉末在具塞试管中容易聚集、不 易在溶剂中分散的性质;3)银杏叶中的咖啡因属于内源性物质。
[0087]
基于上述原因,为了能够检测出银杏叶中的咖啡因,发明人进行了如下处理:
[0088]
1)供试品处理方面:
[0089]
因为咖啡因的含量太少,因此在银杏叶的处理上,就需要除去银杏叶上及内含的杂质, 本发明增加了两个处理:a研成粉末,过四号筛;b、0.2μm针筒式过滤器过滤,取续滤液, 高速离心15min,转数12000转/分钟,取上清液;
[0090]
2)用纯甲醇溶解银杏叶,超声时间为10min,节省了时间,且溶出效果最佳;使用更合 适的具塞锥形瓶,常规方法中银杏叶药材粉末堆积在试管底部,超声提取时,粉末多在下部, 溶剂多在上部,使得每批银杏叶样品与溶剂接触充分程度不一致,导致结果差异性较大。而 用具塞锥形瓶,超声时药材粉末与提取容器接触充分,结果稳定性较好;
[0091]
3)改变进样量与银杏叶中咖啡因含量太少有关,因此需适当增加进样量以达到定
量标准。
[0092]
2、本发明提供的方法,结果更加准确、稳定、可靠。
附图说明:
[0093]
图1-1、1-2、1-3分别为不同浓度甲醇、不同提取时间、不同料液比对银杏叶中咖啡因提 取率的影响;
[0094]
图2-1为现有方法文献2中咖啡因的色谱图;
[0095]
图2-2为甲醇空白溶剂多反应监测色谱图(此图为甲醇空白色谱图);
[0096]
图2-3为咖啡因对照品多反应监测色谱图(此图为咖啡因对照品色谱图);
[0097]
图2-4为银杏叶供试品多反应监测色谱图(此图为银杏叶药材供试品色谱图);
[0098]
图3为线性关系考察结果。
具体实施方式
[0099]
实施例1:一种银杏叶中咖啡因含量的测定方法:
[0100]
1、提取方法
[0101]
(1)供试品溶液的制备
[0102]
取银杏叶样品适量,研成粉末,过四号筛,混匀,称取粉末1.0g,精密称定,置具塞锥形 瓶中,精密加入甲醇10ml,密塞,称重,超声提取10min,放冷,再次称重,用甲醇补足 减失的重量,摇匀,用0.2μm针筒式过滤器过滤,取续滤液,高速离心15min,转数12000 转/分钟,取上清液,备用。
[0103]
(2)对照品溶液的制备
[0104]
取咖啡因对照品2.50mg,精密称定,置于5ml容量瓶中,用甲醇溶解并稀释至刻度, 摇匀,制成浓度为500μg/ml母液,-20℃保存。
[0105]
2、色谱、质谱条件
[0106]
(1)液相色谱条件
[0107]
色谱柱:waters cortecs t3色谱柱(2.1
×
100mm,2.7μm);流动相:a为含0.1%甲酸 水溶液,b为含0.1%甲酸乙腈溶液,梯度洗脱,0~5min,95%a;5~22min,95%~2%a; 22~27min,2%a;27~27.5min,2%~95%a;27.5~32min,95%a。流速:0.3ml/min; 柱温30℃;进样量2μl;
[0108]
其中0.1%甲酸乙腈溶液是将1ml甲酸用1000ml乙腈稀释。
[0109]
(2)质谱参数条件
[0110]
正离子模式;毛细管电压:0.5kv;离子源温度:150℃;雾化温度:500℃;雾化器 流速:1000l/h;多反应监测模式(mrm);离子对195.112
→
138.065(定量)、195.112
→
110.037 (定性);驻留时间分别为0.165s、0.165s;锥孔电压分别为4.0v、4.0v;碰撞能量分别为 18.0v、22.0v
[0111]
3、检测方法:按上述供试品、对照品溶液制备方法分别制备供试品、对照品溶液,注入 液质联用仪按上述色谱、质谱条件进行分析,以咖啡因对照品峰为参照峰,记录峰面积。
[0112]
该方法得到的图,图2-3为咖啡因对照品色谱图,图2-4为银杏叶药材供试品色谱
图。
[0113]
实验例1:方法学考察
[0114]
1、线性关系考察
[0115]
取咖啡因母液500μg/ml,分别稀释5
×
104,1
×
105,2
×
105,5
×
105,1
×
106,5
×
106倍, 按上述色谱、质谱条件进样分析。以咖啡因浓度为横坐标,峰面积为纵坐标,绘制标准曲线, 见图3。
[0116]
图3结果显示:线性方程为y=4004.5x 483.89,r2=0.999。
[0117]
结果表明,咖啡因在0.1ng/ml~10ng/ml范围内,线性关系良好。
[0118]
2、检测限和定量限
[0119]
取梯度稀释的对照品溶液,按上述色谱、质谱条件注入液质联用仪进行分析,以信噪比 等于10(s/n=10)为定量限,测得咖啡因在该方法下定量限为1
×
10-4
ng/ml,即1
×
10-3
μg/kg, 检测限(s/n=3)小于此值。
[0120]
3、精密度试验
[0121]
称取1g银杏叶药材(编号yxy-10),按上述提取方法进行处理,按色谱条件连续进样6 次,以咖啡因对照品峰为参照峰,记录峰面积并计算相对标准偏差(rsd)。
[0122][0123]
其中:
[0124]
s为标准偏差,计算公式为
[0125]
为均值
[0126]
结果见表2中的含量及精密度rsd可接受的范围。
[0127]
表2:样品中待测定成分含量、精密度rsd可接受范围和回收率限度
[0128]
待测定成分含量重复性(rsd%)重现性(rsd%)回收率限度(%)100%1298-10110%1.5395-1021%2492-1050.1%3690-1080.01%4885-11010μg/g(ppm)61180-1151μg/g81675-12010μg/kg(ppb)153270-125
[0129]
表2结果计算rsd值为5.12%。样品中咖啡因含量约为1ng/ml,即10ng/g=10μg/kg, 精密度可接受范围在15%以内。
[0130]
测定结果符合2015年版《中国药典》规定,表明仪器精密度良好。
[0131]
4、重复性试验
[0132]
平行称取6份银杏叶药材(编号yxy-10)各1g,按上述提取方法、色谱条件进行操作, 以咖啡因对照品峰为参照峰,记录峰面积,计算得rsd值为5.87%。
[0133]
测定结果表明:本发明提供的方法重复性较好。
[0134]
5、稳定性试验
[0135]
称取1份银杏叶药材(编号yxy-10)1g,按上述前方法处理后,分别于供试品溶液制 备后的第0、1、2、4、8、12h按上述检测方法进行检测,以咖啡因对照品峰为参照峰,记 录峰面积,计算得rsd值为7.20%。
[0136]
测定结果表明:12h内样品的稳定性较好。
[0137]
6、加样回收率试验
[0138]
平行称取银杏叶药材6份(编号yxy-10)各0.5g,分别加入5ng咖啡因对照品,精密 加入10ml甲醇,提取、检测等均按上述提取、检测方法操作,以咖啡因对照品峰为参照峰, 记录峰面积,计算rsd值。
[0139]
结果见表2(回收率限度)。
[0140]
表2结果显示:计算的rsd值为6.24%,平均加样回收率为83.29%,符合2015年版《中 国药典》规定,表明该方法回收率良好。最低回收率、最高回收率、平均回收率均在要求范 围内。
[0141]
实验例2:样品测定
[0142]
方法学考察项均通过后,可对银杏叶中咖啡因在0.1ng/ml~10ng/ml范围的样品进行定 量研究。
[0143]
1、采用本发明实施例1提供的方法以及bjs 201701方法(不同在于:将原供试品直接 用银杏叶样品替换,其他方法同)检测10批银杏叶样品含量,样品来源于安徽亳州中药材交 易市场。
[0144]
结果见表3
[0145]
表3:样品含量的测定结果
[0146][0147][0148]
表3结果显示:
[0149]
本发明方法检测的银杏叶中咖啡因的含量在0.1526ng/ml~6.1107ng/ml范围内,即银杏 叶中咖啡因的含量1.5260
×
10-6
g/kg~61.1070
×
10-6
g/kg范围内。bjs 201701方法检测的结果 在0.2222~10.2530ng/ml之间,即银杏叶中咖啡因的含量2.2220
×
10-6
g/kg
~102.5300
×
10-6 g/kg范围内。
[0150]
本发明方法,方法学考察结果说明该方法得到的数据准确可靠;而完全按照bjs 201701 方法测定银杏叶中咖啡因的含量,具有咖啡因色谱峰响应低、平行样品检测结果不稳定的缺 点,且本实验仅用外标法对其结果进行简单定量,因此其定量准确性低。
[0151]
综上,采用本发明提供的方法进行检测,结果更加准确、稳定、可靠。
[0152]
2、采用bjs 201701方法结合文献2中的银杏叶提取方法(超高效液相-串联质谱法研究 银杏叶提取物提取过程咖啡因的转移规律(张苹苹等,食品工业科技,2019年22期,34-49), 检测21批银杏叶,样品1-10、11-20分别来源于安徽亳州和河北安国中药材交易市场。
[0153]
检测结果见表4:
[0154]
表4:采用bjs 201701方法检测含量的结果
[0155][0156][0157]
表4结果显示:两个文献结合的方法检测银杏叶中咖啡因含量大致在0.0623-10.2530 ng/ml之间,即银杏叶中咖啡因的含量0.6230
×
10-6
g/kg~102.5300
×
10-6
g/kg范围内,结果具 有咖啡因色谱峰响应低、平行样品检测结果不稳定的缺点,因此定量准确性低。
[0158]
结果表明:采用本发明提供的方法进行检测,结果更加准确、稳定、可靠。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。