1.本发明涉及高效液相色谱技术领域,具体涉及一种丙交酯衍生的手性固定相及其制备方法与应用。
背景技术:
2.众所周知,绝大多数的药物都是由手性分子构成,而两种手性分子可能具有明显不同的生理活性(例如:药效、药物毒性、药物安全性等)。目前世界上销售量名列前茅的药物基本都是手性化合物,可见手性拆分对于制药工业而言非常重要。
3.高效液相色谱(hplc)是常用的手性拆分手段,开发具有高效拆分效果、广泛适用性和高负载量的体系具有十分重要的意义。具有手性拆分能力的体系有很多,包括蛋白质、冠醚、环糊精等天然高分子,但天然高分子一般手性识别能力较低,作为手性固定相(csp)没有实际应用价值,因此,目前商业化的手性拆分材料主要是改性天然高分子(例如:改性的纤维素、改性的壳聚糖等)。然而,虽然现有的许多手性固定相具有广泛的适用性和良好的手性选择性,但是仍存在一些特定结构的手性化合物无法在现有的手性固定相上得到有效的拆分。因此,开发新型的手性固定相具有十分重要的意义。
技术实现要素:
4.本发明的目的在于提供一种丙交酯衍生的手性固定相及其制备方法与应用。
5.本发明所采取的技术方案是:
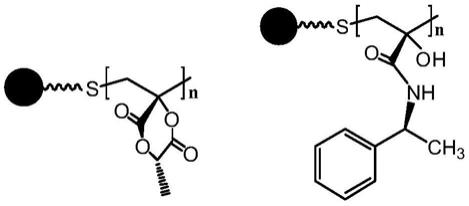
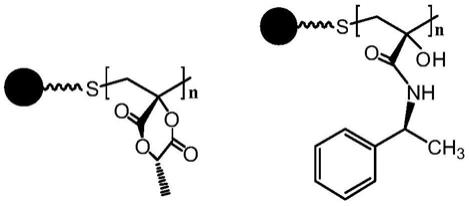
6.一种丙交酯衍生的手性固定相,其结构式如式(i)或式(ii)所示:
[0007][0008]
式(i):sio2‑
pmla,式(ii):sio2‑
pmla
‑
nh2,式中,
●
为硅胶颗粒,n取50~200的整数。
[0009]
式(i)所示的丙交酯衍生的手性固定相的制备方法包括以下步骤:
[0010]
1)进行l
‑
丙交酯和溴代试剂的反应,得到溴代丙交酯
[0011]
2)进行溴代丙交酯和三乙胺的反应,得到亚甲烯丙交酯
[0012]
3)进行硅胶颗粒和巯基硅烷的反应,得到巯基改性硅胶颗粒;
[0013]
4)进行巯基改性硅胶颗粒和亚甲烯丙交酯的反应,得到丙交酯衍生的手性固定相sio2‑
pmla。
[0014]
优选的,式(i)所示的丙交酯衍生的手性固定相的制备方法包括以下步骤:
[0015]
1)将l
‑
丙交酯和溴代试剂分散在溶剂中,再加入偶氮类引发剂,进行反应,得到溴代丙交酯
[0016]
2)将溴代丙交酯分散在溶剂中,再加入三乙胺,进行反应,得到亚甲烯丙交酯
[0017]
3)将硅胶颗粒分散在溶剂中,再加入巯基硅烷,进行反应,得到巯基改性硅胶颗粒;
[0018]
4)将巯基改性硅胶颗粒和亚甲烯丙交酯分散在溶剂中,保护气氛下加入偶氮类引发剂,进行反应,得到丙交酯衍生的手性固定相sio2‑
pmla。
[0019]
优选的,步骤1)所述反应在60℃~80℃下进行,反应时间为15h~24h。
[0020]
优选的,步骤1)所述l
‑
丙交酯、溴代试剂的摩尔比为1:1~1:2。
[0021]
优选的,步骤1)所述溴代试剂为n
‑
溴代琥珀酰亚胺(nbs)、1,3
‑
二溴
‑
5,5
‑
二甲基海因中的至少一种。
[0022]
优选的,步骤1)所述溶剂为甲苯、n,n
‑
二甲基甲酰胺、四氢呋喃中的至少一种。
[0023]
优选的,步骤1)所述偶氮类引发剂为偶氮二异丁腈(aibn)、偶氮二异庚腈(abvn)中的至少一种。
[0024]
优选的,步骤1)所述偶氮类引发剂的添加量为l
‑
丙交酯质量的4%~6%。
[0025]
优选的,步骤2)所述溴代丙交酯、三乙胺的摩尔比为1:1.1~1:1.2。
[0026]
优选的,步骤2)所述溶剂为二氯甲烷、正己烷中的至少一种。
[0027]
优选的,步骤2)所述反应分两阶段进行,第一阶段0℃~4℃反应0.5h~1.5h,第二阶段室温(15℃~35℃)反应0.5h~1.5h。
[0028]
优选的,步骤3)所述反应在110℃~130℃下进行,反应时间为20h~30h。
[0029]
优选的,步骤3)所述硅胶颗粒、巯基硅烷的质量比为1:4~1:6。
[0030]
优选的,步骤3)所述溶剂为甲苯、n,n
‑
二甲基甲酰胺中的至少一种。
[0031]
优选的,步骤3)所述巯基硅烷为3
‑
巯基丙基三甲氧基硅烷(γ
‑
mps)、3
‑
巯基丙基三乙氧基硅烷中的至少一种。
[0032]
优选的,步骤4)所述反应在60℃~80℃下进行,反应时间为20h~30h。
[0033]
优选的,步骤4)所述巯基改性硅胶颗粒、亚甲烯丙交酯的质量比为1:1.2~1:1.6。
[0034]
优选的,步骤4)所述偶氮类引发剂的添加量为亚甲烯丙交酯质量的2%~3%。
[0035]
优选的,步骤4)所述溶剂为1,4
‑
二氧六环、甲苯、n,n
‑
二甲基甲酰胺中的至少一种。
[0036]
优选的,步骤4)所述偶氮类引发剂为偶氮二异丁腈(aibn)、偶氮二异庚腈(abvn)中的至少一种。
[0037]
优选的,步骤4)所述保护气氛为氮气气氛。
[0038]
式(ii)所示的丙交酯衍生的手性固定相的制备方法包括以下步骤:进行sio2‑
pmla和(r)
‑
( )
‑1‑
苯乙胺的反应,即得丙交酯衍生的手性固定相sio2‑
pmla
‑
nh2。
[0039]
优选的,式(ii)所示的丙交酯衍生的手性固定相的制备方法包括以下步骤:将sio2‑
pmla分散在溶剂中,再加入(r)
‑
( )
‑1‑
苯乙胺,进行反应,即得丙交酯衍生的手性固定相sio2‑
pmla
‑
nh2。
[0040]
优选的,所述溶剂为n,n
‑
二甲基甲酰胺(dmf)、四氢呋喃(thf)中的至少一种。
[0041]
优选的,所述反应在室温(15℃~35℃)下进行,反应时间为40h~60h。
[0042]
本发明的有益效果是:本发明的丙交酯衍生的手性固定相具有优异的拆分性能,能够在较低的压力下拆分多种不同类型的对映异构体,分离快速高效,有较好的重现性,可以反复使用,且其制备过程简单、反应条件温和、反应可控,适合进行实际应用。
[0043]
具体来说:
[0044]
1)本发明的sio2‑
pmla
‑
nh2和sio2‑
pmla色谱柱相比于sio2‑
plla色谱柱,具有更加优异的拆分性能,能够在较低的压力下拆分多种不同类型的对映异构体,分离快速高效,有较好的重现性,且可以反复使用;
[0045]
2)本发明的丙交酯衍生的手性固定相的制备方法简单,原料廉价易得,反应条件温和,易于操作,具有良好的市场应用前景。
附图说明
[0046]
图1为硅胶颗粒、实施例1中的sio2‑
pmla和实施例2中的sio2‑
pmla
‑
nh2的sem图。
[0047]
图2为硅胶颗粒、实施例1中的sio2‑
sh、实施例1中的sio2‑
pmla和实施例2中的sio2‑
pmla
‑
nh2的tga曲线。
[0048]
图3为硅胶颗粒、实施例1中的sio2‑
pmla和实施例2中的sio2‑
pmla
‑
nh2的红外光谱图。
[0049]
图4为sio2‑
pmla色谱柱和sio2‑
pmla
‑
nh2色谱柱分离的15种外消旋体的化学结构。
[0050]
图5为sio2‑
pmla色谱柱对15种外消旋体的拆分结果。
[0051]
图6为sio2‑
pmla
‑
nh2色谱柱对15种外消旋体的拆分结果。
[0052]
图7为sio2‑
plla色谱柱对15种外消旋体的拆分结果。
具体实施方式
[0053]
下面结合具体实施例对本发明作进一步的解释和说明。
[0054]
实施例1:
[0055]
一种丙交酯衍生的手性固定相,其制备方法包括以下步骤:
[0056]
1)将20g的l
‑
丙交酯、29.66g的n
‑
溴代琥珀酰亚胺和300ml的甲苯加入带有回流冷凝器的容量500ml的三颈烧瓶中,机械搅拌下将混合物加热回流,再将1.14g的偶氮二异丁腈用50ml的甲苯分散后通过滴液漏斗滴加到三颈烧瓶中,加完后80℃搅拌20h,反应液冷却至室温,过滤,将滤液蒸干,将得到的固体(淡黄色)溶解在二氯甲烷中,用饱和亚硫酸氢钠溶液洗涤3次,用饱和nacl溶液洗涤1次,取有机层用mgso4干燥后蒸干,得到溴代丙交酯(白色晶体,收率53%);
[0057]
2)将20.5g的溴代丙交酯和200ml的二氯甲烷加入容量500ml的三颈烧瓶中,充氮气保护,置于冰浴中冷却,再通过滴液漏斗滴加14.1ml的三乙胺,加完后保持在冰浴中反应1h,再升温至室温搅拌反应1h,将反应液转移至分液漏斗中,用浓度1mol/l的盐酸溶液洗涤3次,用饱和nacl溶液洗涤1次,取有机层用mgso4干燥后蒸干,再通过硅胶柱色谱法纯化,再45℃真空升华,得到亚甲烯丙交酯(白色固体,收率49%);
[0058]
3)将5g的硅胶颗粒(记为sio2)超声分散在50ml的甲苯中,再滴加27ml的3
‑
巯基丙基三甲氧基硅烷(γ
‑
mps),加完后120℃搅拌回流24h,反应液冷却至室温,离心,依次用乙醇、蒸馏水和乙醇洗涤离心得到的固体,60℃真空干燥15h,得到巯基改性硅胶颗粒(记为sio2‑
sh);
[0059]
4)将4.5g的亚甲烯丙交酯和30ml的1,4
‑
二氧六环加入容量100ml的圆底烧瓶中,搅拌分散,再加入3.0g的巯基改性硅胶颗粒,超声分散,充氮气保护,再将130mg的偶氮二异丁腈(aibn)用5ml的1,4
‑
二氧六环分散后加入圆底烧瓶中,70℃搅拌反应24h,反应液冷却至室温,过滤,依次用乙醇、蒸馏水和乙醇洗涤滤得的固体,60℃真空干燥20h,得到丙交酯衍生的手性固定相sio2‑
pmla。
[0060]
sio2‑
pmla的合成反应如下:
[0061][0062]
实施例2:
[0063]
一种丙交酯衍生的手性固定相,其制备方法包括以下步骤:
[0064]
将3.5g实施例1的sio2‑
pmla分散在30ml的无水n,n
‑
二甲基甲酰胺(dmf)中,再滴加18ml的(r)
‑
( )
‑1‑
苯乙胺,加完后室温反应48h,过滤,滤得的固体用乙腈洗涤3次,60℃真空干燥15h,即得丙交酯衍生的手性固定相sio2‑
pmla
‑
nh2。
[0065]
sio2‑
pmla
‑
nh2的合成反应如下:
[0066][0067]
对比例:
[0068]
一种丙交酯衍生的手性固定相,其制备方法包括以下步骤:
[0069]
1)将3.5g的硅胶颗粒超声分散在50ml的甲苯中,再滴加14ml的3
‑
氯丙基三甲氧基硅烷(cptms),加完后氮气保护下120℃搅拌回流48h,反应液冷却至室温,离心,依次用无水甲苯和无水乙醇洗涤离心得到的固体,40℃真空干燥15h,得到氯化改性硅胶颗粒(记为
sio2‑
cl);
[0070]
2)将3.5g的氯化改性硅胶颗粒和7.0g的三羟甲基氨基甲烷加入70ml的n,n
‑
二甲基甲酰胺中,80℃搅拌48h,离心,用水洗涤离心得到的固体多次至中性,再依次用甲醇和丙酮洗涤,40℃真空干燥过夜,得到三羟甲基氨基甲烷改性硅胶颗粒(记为sio2‑
tris);
[0071]
3)将6.0g的l
‑
丙交酯和50ml的无水甲苯加入容量100ml的圆底烧瓶中,再加入3.5g的三羟甲基氨基甲烷改性硅胶颗粒,超声分散,充氮气保护,再将106mg的异辛酸亚锡用5ml的无水甲苯分散后加入圆底烧瓶中,110℃搅拌24h,反应液冷却至室温,过滤,依次用甲苯和氯仿洗涤滤得的固体,60℃真空干燥18h,得到丙交酯衍生的手性固定相sio2‑
plla。
[0072]
sio2‑
plla的合成反应如下:
[0073][0074]
性能测试:
[0075]
1)硅胶颗粒、实施例1中的sio2‑
pmla和实施例2中的sio2‑
pmla
‑
nh2的扫描电镜(sem)图如图1(a为硅胶颗粒,b为sio2‑
pmla,c为sio2‑
pmla
‑
nh2)所示。
[0076]
由图1可知:未经修饰的硅胶颗粒表面光滑,而键合上聚合物后,硅球表面变得粗糙不平,说明亚甲烯丙交酯单体已成功在硅球表面聚合。
[0077]
2)硅胶颗粒、实施例1中的sio2‑
sh、实施例1中的sio2‑
pmla和实施例2中的sio2‑
pmla
‑
nh2的热重分析(tga)曲线如图2所示。
[0078]
由图2可知:sio2‑
sh的失重率约13%,sio2‑
pmla和sio2‑
pmla
‑
nh2的失重率约24%,sio2‑
pmla和sio2‑
pmla
‑
nh2的失重率比sio2‑
sh大,这是因为sio2‑
pmla和sio2‑
pmla
‑
nh2中聚合物的分解,这也表明sio2‑
pmla和sio2‑
pmla
‑
nh2成功制备。
[0079]
3)硅胶颗粒、实施例1中的sio2‑
sh、实施例1中的sio2‑
pmla和实施例2中的sio2‑
pmla
‑
nh2的元素分析结果如下表所示:
[0080]
表1元素分析结果
[0081][0082]
由表1可知:相比于sio2‑
sh,sio2‑
pmla的碳含量从0.68%增加到13.22%,sio2‑
pmla
‑
nh2的碳含量从0.68%增加到14.01%,说明亚甲烯丙交酯成功键合到硅胶颗粒的表面。此外,相比于sio2‑
pmla,sio2‑
pmla
‑
nh2的氮含量从0.01%增加到0.58%,说明对sio2‑
pmla成功进行了胺解改性。
[0083]
4)硅胶颗粒、实施例1中的sio2‑
pmla和实施例2中的sio2‑
pmla
‑
nh2的红外光谱(ft
‑
ir)图如图3所示。
[0084]
由图3可知:1791cm
‑1处为c=o键的伸缩振动峰,说明亚甲烯丙交酯成功键合到硅胶颗粒表面;3435cm
‑1处的峰归属于n
‑
h键的伸缩振动峰,说明对sio2‑
pmla成功进行了胺解改性。
[0085]
5)将3.0g实施例1的sio2‑
pmla或实施例2的sio2‑
pmla
‑
nh2加入15ml的丙酮中,超声分散7min,再迅速将得到的悬浮液倒入匀浆罐中,以丙酮为顶替液将悬浮液压入高效液相色谱柱管中,50mpa压力下装柱5min,再将压力降为0,保压20min,色谱柱的规格为150mm
×
4.6mm(i.d.),再在高效液相色谱上,分离如图4所示的15种外消旋体,反相色谱条件:流动相:ⅰ、纯甲醇;ⅱ、甲醇:水=80:20(v/v);ⅲ、甲醇:水=60:40(v/v);流速:1.0ml/min;柱温:40℃,拆分结果分别如图5和图6所示。
[0086]
由图5可知:在15种外消旋体中,sio2‑
pmla色谱柱可分离7种外消旋体,其中,有4种达到了基线分离。
[0087]
由图6可知:在15种外消旋体中,sio2‑
pmla
‑
nh2色谱柱可分离12种外消旋体,其中,有9种达到了基线分离。
[0088]
由此可见,本发明的丙交酯衍生的手性固定相具有良好的拆分效果,外消旋化合物能够在较短时间内完成高效分离,且sio2‑
pmla
‑
nh2色谱柱的拆分效果更加优异。
[0089]
6)将3.0g对比例的sio2‑
plla加入15ml的丙酮中,超声分散7min,再迅速将得到的悬浮液倒入匀浆罐中,以丙酮为顶替液将悬浮液压入高效液相色谱柱管中,50mpa压力下装柱5min,再将压力降为0,保压20min,色谱柱的规格为150mm
×
4.6mm(i.d.),再在高效液相色谱上,以甲醇作流动相,流速为1.0ml/min,柱温40℃条件下分离如图4所示的15种外消旋体,拆分结果分别如图7所示。
[0090]
由图7可知:在15种外消旋体中,sio2‑
plla色谱柱仅能拆分3种对映异构体,分离效果较差,即使改变分离条件,消旋体也未能得到有效拆分。
[0091]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。