用于wnt六肽的线性溶液相路径
技术领域
1.本公开总体上涉及多肽合成领域,更特别地,涉及对wnt六肽foxy
‑
5的线性溶液相方法。本公开进一步涉及新颖foxy
‑
5三肽、四肽和五肽片段的溶液相合成。
背景技术:
2.foxy
‑
5是甲酰化的、wnt5a衍生的六肽和wnt5a模拟物,具有潜在的抗转移活性,目前正在作为候选药物研发以预防几种常见形式的癌症中的肿瘤扩散。
3.foxy
‑
5具有氨基酸序列for
‑
met
‑
asp
‑
gly
‑
cys
‑
glu
‑
leu
‑
oh(seq_no 1,图1)。
4.静脉内施用后,foxy
‑
5结合并激活wnt5a受体,主要是frizzled家族,frizzled家族激活wnt5a介导的信号传导。
5.foxy
‑
5旨在补偿结肠癌患者中注意到的肿瘤组织中蛋白质wnt5a的缺乏,以降低转移的风险。最近一项对iii期结直肠癌患者的回顾性研究的子分析显示,wnt5a低表达患者的比例明显高于对ii期结直肠癌(crc)患者的先前研究中所观察到的。crc iii期肿瘤患者与ii期患者的不同之处主要在于,在与原发肿瘤相邻的淋巴结中存在肿瘤细胞,因此更具侵袭性且进展更快。在iii期患者的近70%中观察到低水平的wnt5a,相比之下,肿瘤次晚期患者情况下的比例为约45%。这支持了wnt5a水平显著影响疾病进程的假设。
6.基于旨在记录候选药物的安全性概况的已完成的针对foxy
‑
5的1b期研究,针对2期的药代动力学和剂量确定,foxy
‑
5现在用于2期临床试验研究,其中结肠癌患者的治疗将在诊断时在进行手术之前开始。该治疗旨在持续最多12周,或直到开始化疗。
7.foxy
‑
5及其制备方法描述于国际专利号wo06130082 a1。迄今为止,用于临床前和临床研究的活性药物成分(api)是通过经典固相肽合成(spps)生产的,其中foxy
‑
5是通过线性1 1 1 1 1 1路径生产的,见图2。
8.因此,使用fmoc策略(fmoc=芴基甲基氧基羰基)将序列for
‑
met
‑
asp
‑
gly
‑
cys
‑
glu
‑
leu
‑
oh组装在带有c末端氨基酸leu的2
‑
氯三苯甲基树脂上。合成在spps反应器中进行并且由以下组成:交替偶联、乙酰化和n
‑
α
‑
脱保护程序。偶联在作为溶剂的dmf(n,n
‑
二甲基甲酰胺)或dmf/dcm(二氯甲烷)中进行。如果需要的话,它由以下组成:在激活剂和碱的存在下,将n
‑
α
‑
保护的氨基酸衍生物与前面的氨基酸偶联。甲酸在没有激活剂的情况下作为活性酯偶联。
9.如果偶联不完全,则可以继续进行或重复该程序。为了避免形成作为副产物的缺失序列的形成,在偶联步骤之后进行系统性乙酰化步骤(加帽),或者,如果在再偶联步骤之后进行再偶联,则使用dmf、乙酸酐和吡啶。
10.乙酰化之后是n
‑
α
‑
脱保护程序,该程序由以下组成:用dmf洗涤树脂,用在dmf中的哌啶裂解fmoc
‑
基团,并且随后用dmf洗涤。在不完全裂解的情况下,可以重复如上所述的n
‑
α
‑
脱保护程序。对于每个单个步骤,添加溶剂和/或试剂,并且搅拌反应混合物,然后过滤以将溶剂和/或试剂从树脂除去。
11.重复偶联、乙酰化和n
‑
α
‑
脱保护步骤,直到树脂带有完整的肽序列for
‑
met
‑
asp
‑
gly
‑
cys
‑
glu
‑
leu
‑
oh。在甲酸活性酯的最终偶联之后,不进行乙酰化。通过用dmf和ipa洗涤肽树脂并随后在减压下干燥来完成spps。
12.通过在合适的清除剂(例如水和edt)存在下用tfa处理肽树脂来完成从树脂上裂解肽和伴随的侧链保护基团裂解。随后,通过二维制备型hplc在反相柱上使用acn梯度洗脱(甲酸和乙酸系统)纯化获得的粗制肽。
13.将具有足够纯度的合并的级分冻干。在不符合设定的纯度标准的情况下,则将冻干物通过hplc分析,并任选地通过如上概述的二维制备型hplc再纯化。
14.上面概述的spps方法已经为临床前和早期临床研究提供了足够的材料,但是对于进一步的临床研究和最终的商业目的,需要一种更适合大规模合成的合成方法,这样可以降低商品成本并可以提供更大批量的foxy
‑
5。
15.因此,需要一种可靠的合成路径,其可以提供数千克规模的foxy
‑
5,用于进一步的临床试验供应和最终的商业目的。
附图说明
16.图1显示了foxy
‑
5的化学结构。foxy
‑
5是一种线性肽,其由具有甲酰化n末端的六个氨基酸组成。所有光学活性氨基酸残基均为l
‑
构型。foxy
‑
5的分子式为c
26
h
42
n6o
12
s2,并且分子量为694.8g/mol(平均质量)。
17.图2显示了通往foxy
‑
5的spps路径的合成方案。
18.图3说明了形成foxy
‑
5的线性1 1 1 1 1 1策略。
19.图4说明了“天冬酰胺杂质”的形成。
20.图5说明了甲酰基基团在观察到的差向异构化反应中的作用。
21.缩写
22.fmoc=芴基甲氧基羰基
23.tfa=三氟乙酰基
24.tsoc=4
‑
甲苯磺酰基乙基氧基羰基
25.mesoc=甲基磺酰基乙基氧基羰基
26.peoc=2
‑
(三苯基膦酰基)乙基氧基羰基
27.cyoc=2
‑
氰基
‑
叔丁基氧基羰基,以及
28.pht=邻苯二甲酰基
29.nsc=2
‑
(4
‑
硝基苯基磺酰基)乙氧基羰基
30.boc=叔丁基氧基羰基
31.for=甲酰基
32.trt=三苯甲基(trityl)
33.tbu=叔
‑
丁基
34.thf=四氢呋喃
35.dipe=二异丙醚
36.dmf=n,n
‑
二甲基甲酰胺
37.tfa=三氟乙酸
38.tis=三异丙基硅烷
39.hobt=1
‑
羟基苯并三唑
40.dcm=二氯甲烷
41.edac,hcl=1
‑
乙基
‑3‑
(3
′‑
二甲基氨基丙基)碳二亚胺,hcl
42.dipea =二异丙胺
43.dbu=1,8
‑
二氮杂双环[5.4.0]十一
‑7‑
烯
[0044]
氨基酸缩写:
[0045]
met=甲硫氨酸
[0046]
asp=天冬酰胺
[0047]
gly=甘氨酸
[0048]
cys=半胱氨酸
[0049]
glu=谷氨酸
[0050]
leu=亮氨酸
[0051]
foxy
‑
5=for
‑
met
‑
asp
‑
gly
‑
cys
‑
glu
‑
leu
技术实现要素:
[0052]
本公开提供了线性溶液相方法,用于制备被称为foxy
‑
5的甲酰化六肽(即for
‑
met
‑
asp
‑
gly
‑
cys
‑
glu
‑
leu
‑
oh(seq_no 1)),以及其各种三、四、五
‑
及六肽片段,包括其受保护的衍生物。本文提供的方法具有优于传统固相合成的许多优点,包括但不限于低的原料成本,易于纯化工艺中间体,易于片段组装,高的手性纯度以及对商业规模化的适应性,这将在下面更详细地描述。
[0053]
因此,本发明的主要目的是为foxy
‑
5提供可扩展的合成路径。另一个目的是为所述可扩展的合成路径鉴定和表征合适的关键中间体,以用于药物物质的随后gmp(良好生产规范)生产。
[0054]
考虑到通常与固相化学相关的商品成本和繁琐的可扩展性,重点一直放在开发溶液相化学路径上。本公开提供了制备foxy
‑
5或其中间体和前体的线性(1 1 1 1 1 1)溶液相方法。
[0055]
与传统的、收敛式六肽溶液相方法(即先单独产生二肽或三肽,然后再进行偶联)相比,诸位发明人已出人意料地发现,线性路径(即通过氨基酸met、asp、gly、cys、glu和leu的受保护的衍生物的顺序溶液相偶联来组装foxy
‑
5的肽序列)可以使其非常有效地发挥作用。
[0056]
本发明的核心部分在于在甲硫氨酸n
‑
末端上引入甲酰基(for)基团。正如本技术后面将要讨论的,这个特定的化学步骤是最难实现良好的产率和高化学和光学纯度的。
[0057]
诸位发明人现已发现,具有式pg
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or的六肽衍生物可以很好地用作foxy
‑
5前体,并且优选地在当r选自c1‑
c6烷基(如甲基或叔丁基),并且pg是氮的碱敏感性保护基团(即在酸性条件下稳定但在碱性/碱性条件下可从肽上裂解的保护基团)时。在本发明的上下文中,保护基团如fmoc、tfa、tsoc、mesoc、peoc、cyoc或nsc是合适的,优选fmoc。
[0058]
在第一方面,本发明因此提供了具有式pg
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or的六肽衍生物,其中r选自c1‑
c6烷基,如甲基或叔丁基,并且pg是碱敏感性保
护基团,如fmoc、tfa、tsoc、mesoc、peoc、cyoc或nsc。
[0059]
通过n
‑
脱保护,然后与甲酸偶联,并且最后将剩余的保护基团整体脱保护,可以将第一方面的六肽衍生物转化为所期望的六肽foxy
‑
5。
[0060]
在第二方面,本发明因此提供了用于制备六肽foxy
‑
5(seq_no 1)的方法,该方法包括:
[0061]
a.提供如第一方面所述的六肽衍生物,
[0062]
b.从所述六肽衍生物中除去pg保护基团,
[0063]
c.将步骤b)获得的产物与甲酸或其活性酯偶联,以产生受保护的foxy
‑
5衍生物for
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or,
[0064]
d.将步骤c)获得的受保护的foxy
‑
5衍生物整体脱保护以产生以粗品形式的foxy
‑
5,
[0065]
e.任选地进行另外的纯化步骤,以及
[0066]
f.任选地,沉淀形成的呈固体形式的呈碱性或酸性盐的foxy
‑
5六肽,
[0067]
其中r选自c1‑
c6烷基,如甲基或叔丁基,并且pg是碱敏感性保护基团,如fmoc、tfa、tsoc、mesoc、peoc、cyoc或nsc。
[0068]
可以通过任何方便的方法(如固相合成或溶液相方法),并且最方便地通过由下文提出的诸位发明人发展的方法制备第一方面的六肽衍生物。
[0069]
在第三方面,本发明提供了用于制备如第一方面所述的六肽衍生物的方法,该方法包括以下步骤:
[0070]
a.提供受保护的l
‑
亮氨酸衍生物pg
‑
leu
‑
or,
[0071]
b.从所述受保护的l
‑
亮氨酸衍生物中除去保护基团pg,然后与pg
‑
glu
‑
otbu偶联以产生受保护的二肽pg
‑
glu(otbu)
‑
leu
‑
or,
[0072]
c.从所述所述受保护的二肽中除去保护基团pg,然后与pg
‑
cys(trt)
‑
oh偶联以产生受保护的三肽pg
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or,
[0073]
d.从所述受保护的三肽中除去保护基团pg,然后与pg
‑
gly
‑
oh偶联以产生受保护的四肽pg
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or,
[0074]
e.在碱性条件下,从所述受保护的四肽中除去保护基团pg,然后除去过量的碱,以产生去封闭的四肽h
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or,
[0075]
f.在碱性条件下,将所述去封闭的四肽与pg
‑
asp(otbu)偶联,然后除去过量的碱,以产生受保护的五肽pg
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or,
[0076]
g.从所述受去封闭的五肽中除去保护基团pg,然后与甲酸或其活性酯偶联以产生受保护的六肽pg
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or,
[0077]
其中r选自c1‑
c6烷基,如甲基或叔丁基,并且pg是碱敏感性保护基团,如fmoc、tfa、tsoc、mesoc、peoc、cyoc或nsc。
[0078]
下面将进一步讨论本发明的上述方面(包括其优选的实施方案)。
[0079]
在本发明的第四方面,提供了基于三肽中间体intm
‑
2(fmoc
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu)与氨基酸gly、asp和met的受保护的衍生物的顺序偶联,然后n
‑
脱保护并且与甲酸偶联来产生受保护形式的六肽foxy
‑
5的方法。
[0080]
在本发明的第五方面,提供了基于四肽中间体intm
‑
3(fmoc
‑
gly
‑
cys(trt)glu
(otbu)
‑
leu
‑
otbu)与氨基酸asp和met的受保护的衍生物的顺序偶联,然后n
‑
脱保护并且与甲酸偶联来产生受保护形式的六肽foxy
‑
5的方法。
[0081]
在本发明的另外方面,提供了以下肽:
[0082]
fmoc
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu
[0083]
fmoc
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu(seq_no 2)
[0084]
fmoc
‑
asp(otbu)
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu(seq_no 3)
[0085]
fmoc
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu(seq_no 4)
[0086]
for
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu(seq_no 5)
[0087]
cys
‑
glu
‑
leu
[0088]
gly
‑
cys
‑
glu
‑
leu(seq_no 6)
[0089]
asp
‑
gly
‑
cys
‑
glu
‑
leu(seq_no 7)
[0090]
met
‑
asp
‑
gly
‑
cys
‑
glu
‑
leu(seq_no 8)
具体实施方式
[0091]
如上文发明内容中所述,设计了用于组装foxy
‑
5六肽序列的线性溶液相方法,这将在下文进行更详细的讨论。
[0092]
一般的方法是保护所有的氨基酸作为o
‑
t
bu、n
‑
fmoc衍生物。此外,目标是尽可能“简略”的合成,从而避免中间体的耗时和昂贵的分离。优选地,序列中的每个步骤应该可以在没有产物分离的情况下进行,只允许进行小的纯化步骤,如有机溶剂溶液的水性后处理和硅胶塞处理(“快速色谱法”)以除去例如过量的碱。
[0093]
线性合成开始于在两个简略的步骤(步骤1 2)中,通过将叔丁基l
‑
亮氨酸酯,hcl与fmoc
‑
glu(otbu)在作为反应溶剂的二氯甲烷(dcm)中偶联,制备三肽intm
‑
2(fmoc
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu)。将反应混合物用水和盐水处理,并且将dcm溶液直接用于步骤2,而不分离中间二肽,intm
‑
1(fmoc
‑
glu(otbu)leu
‑
otbu)。
[0094]
在步骤2中,将intm
‑
1的dcm溶液与fmoc
‑
cys(trt)
‑
oh反应,以得到所期望的三肽intm
‑
2(fmoc
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu)。反应进行得很好,并且经两步重复给出约68%的合并产率。可以将intm
‑
2通过硅胶(100
‑
200)色谱法纯化为白色固体。获得的dcm溶液也可以照原样向下进行,并将其直接用于步骤3,与fmoc
‑
gly
‑
oh偶联。
[0095]
在步骤3中,将intm
‑
2用dbu进行fmoc
‑
脱保护,并且在dcm中与fmoc
‑
gly
‑
oh偶联,以得到四肽intm
‑
3(fmoc
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu)。反应完成后,将dcm层用水和盐水洗涤。然后将最终的有机层从50体积浓缩至10
‑
15体积,并照原样向下进行至下一阶段(步骤4)而不分离。
[0096]
在步骤4中,首先将intm
‑
3的dcm溶液用dbu处理以实现fmoc基团的脱保护。去封闭后,在将四肽在edc.hcl、hobt水合物和dipea的存在下与fmoc
‑
asp
‑
otbu偶联之前,将反应物质通过二氧化硅塞柱以除去dbu。反应完成后,将反应物质再次通过二氧化硅塞柱以除去仍存在的dipea。如所述的dbu和dipea的去除对于抑制不期望的天冬酰胺杂质的形成至关重要(参见图4)。以这种方式进行,在100
‑
160克的规模下,经两步(步骤3 4)观察到intm
‑
4(fmoc
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
otbu)的产率为约60%。在dbu的存在下,观察到的产率仅为约25%。
[0097]
在步骤5中,将intm
‑
4在作为溶剂的dcm中用dbu进行fmoc
‑
脱保护,并且与fmoc
‑
甲硫氨酸偶联,以得到以粗品形式的中间体intm
‑
5(fmoc
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
otbu(seq_no 4))。通过柱色谱法使用dcm/thf作为洗脱剂进行纯化。将纯化的产物在dipe中制浆,以得到白色固体。
[0098]
最初,试图将intm
‑
4与n
‑
甲酰基
‑
甲硫氨酸(for
‑
met
‑
oh)直接偶联以产生六肽intm
‑
6,但发现这种合成策略导致最终产物中的部分差向异构化,这是因为for
‑
met
‑
oh在反应条件下可逆地环化,产生噁唑烷酮,这种可逆地环化导致for
‑
met
‑
oh试剂外消旋。参见图5。相反,使用fmoc
‑
甲硫氨酸,然后对fmoc基团进行dbu
‑
脱保护,并且与甲酸或其活性酯偶联,得到所期望的中间体六肽intm
‑
6。于此(trt和o
‑
tbu基团的)整体脱保护提供以粗品形式的foxy
‑
5,可以将该以粗品形式的foxy
‑
5进一步纯化(例如,通过色谱法),和/或作为固体(如酸性或碱性加成盐)沉淀。
[0099]
在第一方面,本发明因此提供了具有式pg
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or的六肽衍生物,其中r选自c1‑
c6烷基,如甲基或叔丁基,并且pg是碱敏感性保护基团,如fmoc、tfa、tsoc、mesoc、peoc、cyoc或nsc。
[0100]
在第一方面的实施方案中,pg是fmoc。
[0101]
在第一方面的另一个实施方案中,提供了具有下式的六肽衍生物:fmoc
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or,其中r选自c1‑
c6烷基,如甲基或叔丁基。
[0102]
在第一方面的优选的实施方案中,六肽衍生物具有式fmoc
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
otbu(seq_no 4)。
[0103]
在第二方面,本发明提供了用于制备六肽foxy
‑
5(seq_no 1)的方法,该方法包括:
[0104]
a.提供如第一方面所述的六肽衍生物,
[0105]
b.从所述六肽衍生物中除去pg保护基团,
[0106]
c.将步骤b)获得的产物与甲酸或其活性酯偶联,以产生受保护的foxy
‑
5衍生物for
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or,
[0107]
d.将步骤c)获得的受保护的foxy
‑
5衍生物整体脱保护以产生以粗品形式的foxy
‑
5,
[0108]
e.任选地进行另外的纯化步骤,以及
[0109]
f.任选地,沉淀形成的呈固体形式的呈碱性或酸性盐的foxy
‑
5六肽,
[0110]
其中r选自c1‑
c6烷基,如甲基或叔丁基,并且pg是碱敏感性保护基团,如fmoc、tfa、tsoc、mesoc、peoc、cyoc或nsc。
[0111]
在第二方面的实施方案中,pg是fmoc。
[0112]
在第二方面的另一个实施方案中,烷基基团r是叔丁基。
[0113]
在第二方面的另一个实施方案中,与甲酸或其活性酯的偶联在溶液中发生。
[0114]
在第二方面的另一个实施方案中,将获得的粗foxy
‑
5通过色谱法(如反相色谱法)纯化。
[0115]
在第二方面的另一个实施方案中,将获得的foxy
‑
5作为呈固体形式的碱性或酸性盐沉淀。
[0116]
在第二方面的另一个实施方案中,将获得的foxy
‑
5作为晶体碱性或酸性盐分离。
[0117]
在第三方面,本发明提供了用于制备如第一方面所述的六肽衍生物的方法,该方
法包括以下步骤:
[0118]
a.提供受保护的l
‑
亮氨酸衍生物pg
‑
leu
‑
or,
[0119]
b.从所述受保护的l
‑
亮氨酸衍生物中除去保护基团pg,然后与pg
‑
glu
‑
otbu偶联以产生受保护的二肽pg
‑
glu(otbu)
‑
leu
‑
or,
[0120]
c.从所述所述受保护的二肽中除去保护基团pg,然后与pg
‑
cys(trt)
‑
oh偶联以产生受保护的三肽pg
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or,
[0121]
d.从所述受保护的三肽中除去保护基团pg,然后与pg
‑
gly
‑
oh偶联以产生受保护的四肽pg
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or,
[0122]
e.在碱性条件下,从所述受保护的四肽中除去保护基团pg,然后除去过量的碱,以产生去封闭的四肽h
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or,
[0123]
f.在碱性条件下,将所述去封闭的四肽与pg
‑
asp(otbu)偶联,然后除去过量的碱,以产生受保护的五肽pg
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or,
[0124]
g.从所述受去封闭的五肽中除去保护基团pg,然后与甲酸或其活性酯偶联以产生受保护的六肽pg
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
or,
[0125]
其中r选自c1‑
c6烷基,如甲基或叔丁基,并且pg是碱敏感性保护基团,如fmoc、tfa、tsoc、mesoc、peoc、cyoc或nsc,
[0126]
其中r选自c1‑
c6烷基,如甲基或叔丁基,并且pg是碱敏感性保护基团,如fmoc、tfa、tsoc、mesoc、peoc、cyoc或nsc。
[0127]
在第三方面的实施方案中,pg是fmoc。
[0128]
在第三方面的实施方案中,所有的步骤b)
‑
g)都在溶液中进行。
[0129]
在另外的实施方案中,可通过如第三方面所述的方法获得如第一方面所述的六肽衍生物。
[0130]
在第三方面的优选的实施方案中,碱敏感性保护基团pg是fmoc,并且烷基基团r是叔丁基。
[0131]
在第三方面的另一个实施方案中,在没有产物分离的情况下进行至少两个连续的偶联步骤,如两个、三个或四个步骤。
[0132]
在优选的实施方案中,本发明因此提供了用于产生呈粗品形式的六肽foxy
‑
5的以下步骤顺序:
[0133]
1.将leu
‑
otbu与fmoc
‑
glu
‑
otbu偶联以产生二肽intm
‑
1(fmoc
‑
glu(otbu)
‑
leu
‑
otbu),然后
[0134]
2.于此与fmoc
‑
cys(trt)
‑
oh偶联以产生三肽intm
‑
2(fmoc
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu),然后
[0135]
3.于此与fmoc
‑
gly
‑
oh偶联以得到受保护的四肽intm
‑
3(fmoc
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu(seq_no 2)),然后
[0136]
4.于此与fmoc
‑
asp
‑
otbu偶联以得到受保护的五肽intm
‑
4(fmoc
‑
asp(otbu)
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu(seq_no 3)),然后
[0137]
5.与fmoc
‑
met偶联以得到受保护的六肽intm
‑
5(fmoc
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu(seq_no 4)),然后
[0138]
6.于此与甲酸反应以得到呈受保护形式的foxy
‑
5,即intm
‑
6(for
‑
met
‑
asp
(otbu)
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu(seq_no 5)),然后
[0139]
7.将t
‑
bu和trt基团整体脱保护以产生呈粗品形式的foxy
‑
5,即for
‑
met
‑
asp
‑
gly
‑
cys
‑
glu
‑
leu
‑
oh(seq_no 1)。
[0140]
在第四方面,提供了基于三肽中间体intm
‑
2(fmoc
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu)与氨基酸gly、asp和met的受保护的衍生物的顺序偶联,然后n
‑
脱保护并且与甲酸偶联来产生受保护形式的六肽foxy
‑
5(即intm
‑
6(for
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu(seq_no 5)))的方法。
[0141]
在实施方案中,通过固相合成产生中间体intm
‑
2(fmoc
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu)。在优选的实施方案中,通过溶液相合成产生所述中间体intm
‑
2。
[0142]
在第五方面,提供了基于四肽中间体intm
‑
3(fmoc
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu)与氨基酸asp和met的受保护的衍生物的顺序偶联,然后n
‑
脱保护并且与甲酸偶联来产生受保护形式的六肽foxy
‑
5(即intm
‑
6(for
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu(seq_no 5)))的方法。
[0143]
在实施方案中,通过固相合成产生中间体intm
‑
3(fmoc
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu)。在优选的实施方案中,通过溶液相合成产生所述中间体intm
‑
3。
[0144]
在本发明的另外方面,提供了以下肽:
[0145]
fmoc
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu
[0146]
fmoc
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu(seq_no 2)
[0147]
fmoc
‑
asp(otbu)
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu(seq_no 3)
[0148]
fmoc
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu(seq_no 4)
[0149]
for
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu(seq_no 5)
[0150]
cys
‑
glu
‑
leu
[0151]
gly
‑
cys
‑
glu
‑
leu(seq_no 6)
[0152]
asp
‑
gly
‑
cys
‑
glu
‑
leu(seq_no 7)
[0153]
met
‑
asp
‑
gly
‑
cys
‑
glu
‑
leu(seq_no 8)
[0154]
实验
[0155]
实施例1三肽intm
‑
2(fmoc
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu),步骤1 2
[0156]
在edac,hcl(2.0当量)、和hobt(2.0当量)和dipea(5.0当量)的存在下,在2025ml二氯甲烷(27体积)和225ml thf(3体积)的溶剂混合物中将50克叔丁基亮氨酸酯.hcl在n2下与175克(2.0当量)fmoc
‑
glu
‑
(otbu)偶联,初始在0℃
‑
5℃下持续1小时,然后在15℃
‑
20℃下持续1小时,得到二肽intm
‑
1(fmoc
‑
glu
‑
(otbu)
‑
leu
‑
otbu)。通过1h nmr和质谱确认身份。为了在下一步中与fmoc
‑
cys(trt)
‑
oh反应,省略了产物分离,并且在水性后处理后直接使用二氯甲烷溶液。因此,使用上述二氯甲烷溶液进行获得的二肽intm
‑
1(fmoc
‑
glu
‑
(otbu)
‑
leu
‑
otbu)的后续反应。使用dbu实现fmoc脱保护,并在edac,hcl(2.0当量)、hobt(2.0当量)和dipea(4当量)存在下与204克(1.1当量)fmoc
‑
cys(trt)
‑
oh偶联,得到粗三肽intm
‑
2,将其通过硅胶(100
‑
200)色谱法纯化,使用etoac
‑
己烷作为洗脱剂,以得到呈灰白色固体的三肽intm
‑
2(fmoc
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu)(148克,经两次偶联反应的总产率为68%,通过hplc纯度为93.8%)。
[0157]
从75克叔丁基亮氨酸酯.hcl重复上述伸缩反应步骤1 2,以得到208克intm
‑
2。
[0158]
实施例2
‑
四肽(fmoc
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu),步骤3
[0159]
将上文获得的intm
‑
2(fmoc
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu)与dbu在dcm(50体积)中反应以实现fmoc脱保护,并且在dipea(3当量)、edc.hcl(2.0当量)和hobt(2.0当量)存在下与1.3当量fmoc
‑
gly
‑
oh反应,以得到受保护的四肽intm
‑
3(fmoc
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
otbu(seq_no 2))。通过tlc观察到良好的转化,并且用水和盐水洗涤dcm层。将最终的有机层浓缩至10
‑
15体积,并照原样用于下一步而不分离。
[0160]
实施例3
‑
五肽(fmoc
‑
asp(otbu)
‑
gly
‑
cys(trt)glu(otbu)
‑
leu
‑
otbu),步骤4
[0161]
如上文中获得的作为浓缩的dcm溶液的关键中间体intm
‑
3(fmoc
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
otbu)首先与dbu反应以实现fmoc脱保护。在进行下一偶联步骤之前,使反应物质通过二氧化硅塞以除去dbu,dbu在先前的实验中已发现可诱导形成不期望的天冬酰胺副产物。在与fmoc
‑
asp
‑
otbu偶联之前去除dbu可有效抑制天冬酰胺形成。硅胶塞处理后,将dcm溶液在dipea、edac,hcl(1.2当量)和hobt(1.2当量)存在下与fmoc
‑
asp(otbu)反应,以得到受保护的五肽intm
‑
4(fmoc
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
otbu(seq_no 3))。通过1h nmr和质谱法确认产物身份。
[0162]
从148克和190克intm
‑
2开始,重复两次伸缩反应(步骤3 步骤4),分别得到107克和178克intm
‑
4(理论值的58.1%和75.4%)。
[0163]
实施例4
‑
六肽(fmoc
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
otbu),步骤5
[0164]
用dbu实现从实施例3获得的五肽intm
‑
4的fmoc脱保护,并在dipea(3.0当量)、edc.hcl(2.0当量)和hobt.h2o(2.0当量)存在下,与fmoc
‑
met在作为溶剂的dcm
‑
thf(50体积 10体积)中偶联,得到呈粗品形式的受保护的foxy
‑
5衍生物intm
‑
5(fmoc
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
otbu(seq_no 4))。通过柱色谱法使用dcm/thf作为洗脱剂进行纯化。将纯化的产物在dipe中制浆,以得到白色固体。
[0165]
纯化在各种条件下进行几次,例如用抗溶剂沉淀和色谱法。发现最好的解决方案是柱色谱法,然后在dipe中制浆,以25克的规模给出78%的产率和95.4%的化学纯度。将反应以80克的规模重复以提供74克化学纯度为94.1%的产物(83%产率)。
[0166]
实施例5
‑
六肽intm
‑
6(for
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
otbu),步骤6
[0167]
用在dcm(50体积)中的dbu实现了实施例4中获得的六肽intm
‑
5的fmoc脱保护,然后在edc.hcl(4.0当量)、hobt.h2o(4.0当量)和dipea(4.0当量)的存在下与甲酸(3.0当量)偶联,以产生intm
‑
6(for
‑
met
‑
asp(otbu)
‑
gly
‑
cys(trt)
‑
glu(otbu)
‑
leu
‑
otbu(seq_id no 5))。
[0168]
将反应(步骤6)分别从4克、18克和18克intm
‑
5进行三次,以得到75%
‑
83%的产率和67.5%
‑
77.2%之间的化学纯度。将反应以30克的规模用10当量甲酸重复以得到17克化学纯度为88.8%的产物(67%产率)。
[0169]
实施例6
‑
六肽for
‑
met
‑
asp
‑
gly
‑
cys
‑
glu
‑
leu
‑
oh(foxy
‑
5),步骤7
[0170]
将实施例5获得的17克六肽通过在tfa(10体积)/(i
‑
pr)3sih(tis,1.7体积)/dtt(1.7当量)的混合物中在n2下在10℃
‑
15℃下溶解和搅拌15
‑
30min而整体脱保护(trt和tbu基团)。接着,将反应混合物温热至25℃至30℃,并且在这个温度下搅拌1至2小时。然后将反
应物质在减压下浓缩至2至3体积。反应完成后,添加thf(5体积),并且在25℃
‑
30℃下再继续搅拌10
‑
15min。然后缓慢添加mtbe(30体积)以沉淀粗产物,其以定量产率(12.3克)以固体获得。
[0171]
将粗产物最后通过反相色谱法使用以下方法条件纯化,得到纯度为98.3%的所期望的六肽for
‑
met
‑
asp
‑
gly
‑
cys
‑
glu
‑
leu
‑
oh(foxy
‑
5):
[0172]
介质:luna c18(3)(品牌:phenomenex)孔径:10μm
[0173]
柱id:50mm id x 250mm(诺华赛公司(novasep)),流速:50.0ml/分钟。
[0174]
样品制备:将100g样品溶解于4ml稀释剂中,通过0.45μm过滤器过滤。
[0175]
缓冲液制备:
[0176]
缓冲液
‑
a:通过将10ml的三氟乙酸混合到10l纯净水中,制备在水中的0.10%
±
0.01%三氟乙酸缓冲液。
[0177]
缓冲液
‑
b:通过将5ml的三氟乙酸混合到5l乙腈中,制备在乙腈中的0.10%
±
0.01%三氟乙酸缓冲液。
[0178]
操作程序:
[0179]
1.将柱用[缓冲液a:缓冲液b]以比例(95:5)平衡3
‑
5个柱体积,流速为5.0ml/min。
[0180]
2.将样品溶液上样到柱上。
[0181]
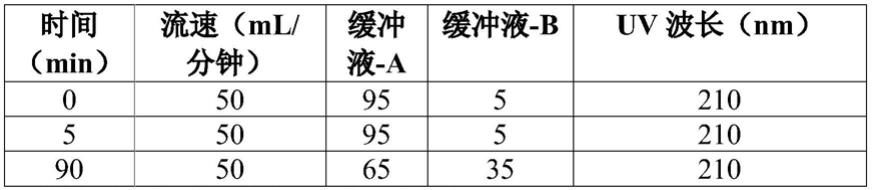
3.对连接到柱上的色谱系统设定程序,以提供如下的梯度程序来开始产物洗脱。
[0182][0183]
4.收集以下级分:前部、顶点和尾部
[0184]
注释:由于规模依赖性,洗脱时间可能因运行而有所不同。
[0185]
5.峰洗脱后,将柱立即用水和乙腈以20:80(%v/v)的比例清洗2个柱体积。
[0186]
6.将级分送去进行纯度分析。
[0187]
7.在
‑
20℃
±
2℃下存储级分。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。