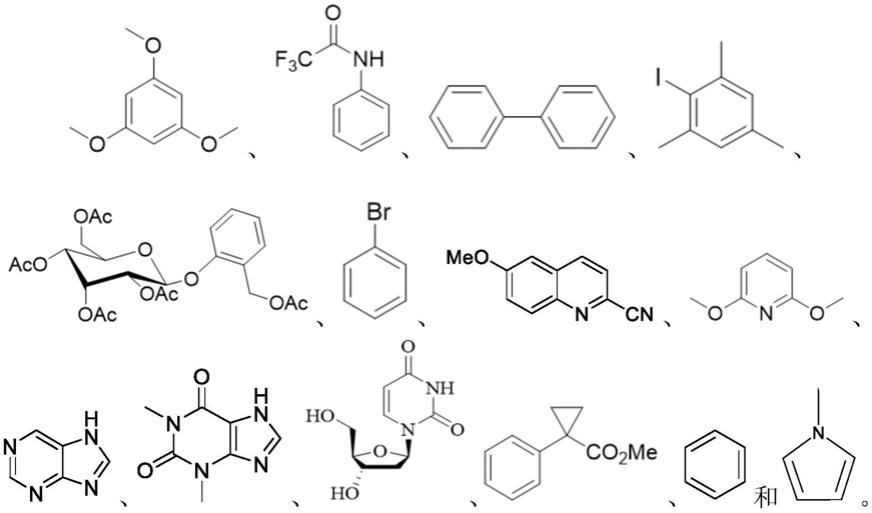
水溶性维生素e参与的共轭二炔类化合物的绿色制备方法
技术领域
1.本发明涉及一种水溶性维生素e参与的共轭二炔类化合物的绿色合成方法。
背景技术:
2.1,3
‑
二炔类化合物由于具有刚性结构单元与独特的电子特性而在药物分子的合成、天然产物的合成以及导电等方面都具有广泛的应用。除此之外,1,3
‑
二炔类化合物在超分子轮烯、超分子开关以及金属螯合剂方面也扮演着十分重要的角色。同时,该类化合物在抗真菌、抗病毒、抗癌症、抗艾滋病等方面都有广泛的生理活性。
3.1,3二炔类化合物的合成方法很多,其中卡尔
·
格拉泽于1869年首次发现了一种经典的、有效的制备双炔类化合物的方法,该方法采用亚铜盐(cucl)两步合成:(1)苯乙炔首先和氯化亚铜在氨和乙醇的混合溶剂中形成苯基乙炔基铜;(2)随后将苯基乙炔铜分离出来,在空气氧化的条件下偶联生成1,4
‑
二苯基
‑
1,3
‑
丁二炔,这就是glaser偶联反应(反应式如下)。这个反应期间要先把苯基乙炔铜分离出来,但是因为其稳定性差、易爆炸、难分离,该方法没有被广泛应用。
4.随后的研究表明,各种铜盐可使末端炔烃发生氧化同系偶联反应。2005年,najera课题组以非磷配体二吡啶
‑2‑
甲基胺衍生物和氯化钯合成配体为催化剂,端炔类化合物可以在n
‑
甲基吡咯烷酮(nmp)溶剂中顺利偶联产生目标产物。
[0005][0006]
这些反应通常在有机溶剂中进行,如甲醇、乙内酯、吡啶、甲基纤维素、苯和甲苯。这些溶剂往往会带来与废物处理相关的问题。
[0007]
本发明以水为溶剂,在催化剂与水溶性维生素e参与的作用下高效的合成共轭二炔类化合物,提供了一种在水相中合成1,3
‑
二炔类化合物的绿色合成方法。
技术实现要素:
[0008]
为了解决现有技术安全性差、废物多等问题,本发明提供一种式ii所示的共轭二端类化合物绿色简便的合成方法,该方法在水溶性维生素e tpgs
‑
750
‑
m形成的微胶束参与下,在水相中完成了共轭二炔类化合物的合成,该发明不仅具有反应底物适用性广的特点,而且减少了有机溶剂的使用,避免了传统方法的条件苛刻,环境危害严重等缺陷,具有反应条件温和、高效、绿色环保等特点,响应了可持续发展理念,实现了溶剂零排放。
[0009]
为了实现上述目的,本发明采用如下技术方案:
[0010]
本发明提供了一种式(ii)所示的共轭二炔类化合物的绿色合成方法,所述方法为:
[0011]
以式(i)所示的取代炔烃为底物,在催化剂与碱性物质的共同作用下,在含1wt%
‑
5wt%(优选2wt%)表面活性剂的水溶液中15
‑
30℃下反应6
‑
15h(优选室温下反应8小时),所得反应液经后处理,得到所述的式(ii)所示的共轭二炔类化合物;所述式(i)所示的取代炔烃、催化剂与碱性物质的物质的量之比为1:0.1
‑
0.3:0.5
‑
2(优选1:0.2
‑
0.3:1);
[0012][0013]
其中,r选自下列基团之一:苯基、卤素取代的苯基、c1~c5的烷基取代的苯基、c1~c5烷氧基取代的苯基、苯甲基、噻吩、三元碳环(如环丙基)、c1~c8的烷基、异丙醇基;
[0014]
所述催化剂选自下列物质之一:硫酸铜(可以五水合硫酸铜的形式加入)、氯化铜、氯化亚铜、醋酸铜(可以一水合醋酸铜的形式加入)(优选为醋酸铜);所述的碱性物质选自下列之一:氢氧化钠、三乙胺、哌啶(优选为哌啶);所述表面活性剂为tpgs
‑
750
‑
m。(碱性物质为碳酸钾时,几乎没有产物生成)
[0015]
反应式如下:
[0016][0017]
本发明推荐所述反应在搅拌条件下进行。优选所述式(i)所示的取代炔烃、催化剂与碱性物质在含表面活性剂的水溶液中搅拌均匀后再进行反应。
[0018]
优选地,所述式(i)所示的取代炔烃为下列化合物之一:
[0019][0020]
进一步,所述表面活性剂的水溶液的体积以式(i)所示的取代炔烃的物质的量计为2
‑
5ml/mmol(优选3.3ml/mmol)。
[0021]
进一步,所述后处理为:向所述反应液中加入乙酸乙酯,搅拌3
‑
8分钟(优选5分钟)后,分离出有机相并进行真空减压下除去溶剂后得到式(ii)所示的共轭二炔类化合物。
[0022]
具体地,所述tpgs
‑
750
‑
m按如下方法制备:
[0023]
(1)搅拌条件下,向生育酚和琥珀酸酐的甲苯溶液中加入三乙胺。随后继续搅拌。反应结束后,向反应混合物中加入水,然后用二氯甲烷萃取。合并的有机层用1mol/l的盐酸和水洗涤,用无水硫酸钠干燥,并在真空减压下除去溶剂,得到黄色液体,将其使用硅胶柱色谱法纯化。洗脱液经真空减压下除去溶剂后得到呈白色固体状的生育酚琥珀酸酯;
[0024]
(2)将含有生育酚琥珀酸酯,聚乙二醇单甲醚
‑
750和对甲苯磺酸的甲苯混合物使用迪安
‑
斯达克榻分水器回流。反应结束后,冷却至室温,将混合物倒入饱和碳酸氢钠水溶液中,并用二氯甲烷萃取。合并的有机层用饱和碳酸氢钠,饱和氯化钠洗涤,用无水硫酸钠干燥,真空减压下除去溶剂后得到目标产物。
[0025]
与现有技术相比,本发明的有益效果在于:以水作为反应溶剂,减少了有机溶剂的使用量,实现了溶剂的零排放;充分利用水的优秀理化性质,反应条件温和且高效,表面活性剂tpgs
‑
750
‑
m通过处理可以回收利用,完全符合环境友好的原则;反应底物适用性广,可以催化芳基炔烃的同时也可以催化脂肪炔烃,为合成共轭二炔提供了一个简易环保的制备方法;筛选出了更适用于本发明反应和反应介质的铜催化剂,大大提高了产率。
[0026]
说明书附图
[0027]
图1是水溶性维生素e(tpgs
‑
750
‑
m)的1h nmr谱;
[0028]
图2是水溶性维生素e(tpgs
‑
750
‑
m)的
13
c nmr谱。
具体实施方式
[0029]
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
[0030]
进一步,本发明使用的表面活性剂十二烷基硫酸钠(sds)、聚乙二醇辛基苯基醚(trition x
‑
100)为市售产品,生育酚甲氧基聚乙二醇琥珀酸溶液(tpgs
‑
750
‑
m,水溶性维生素e)可以购买获得,但本发明的tpgs
‑
750
‑
m为自制,优选为2wt.%tpgs
‑
750
‑
m。
[0031]
实施例中使用的表面活性剂tpgs
‑
750
‑
m为自制,制备方法如下:
[0032][0033]
第一步:向搅拌下的生育酚(4.3g,10mmol)和琥珀酸酐(1.5g,15mmol)的甲苯(20ml)溶液中加入三乙胺(0.35ml,2.5mmol)。随后,在60℃下继续搅拌5h。反应结束后,向反应混合物中加入水(10ml),然后用二氯甲烷(3
×
10ml)萃取。合并的有机层用1mol/l的盐酸(3
×
50ml)和水(2
×
30ml)洗涤,用无水硫酸钠干燥,并真空浓缩,得到生育酚琥珀酸酯粗品,最后利用重结晶方法得到呈白色固体状的生育酚琥珀酸酯(5.25g,收率99%)。结晶条件为:结晶时间26小时,结晶温度4℃,溶剂为正己烷,正己烷/生育酚琥珀酸酯粗品的用量8ml/g。
[0034]
第二步:将含有生育酚琥珀酸酯(2.97g,5.6mmol),聚乙二醇单甲醚
‑
750(4g,5.33mmol)和对甲苯磺酸(0.15g,0.79mmol)的甲苯(20ml)混合物使用迪安
‑
斯达克榻分水器回流5小时。反应结束后,冷却至室温,将混合物倒入饱和碳酸氢钠水溶液中,并用二氯甲烷(3
×
10ml)萃取。合并的有机层用饱和碳酸氢钠(3
×
50ml),饱和氯化钠(2
×
30ml)洗涤,用无水硫酸钠干燥,真空减压下除去溶剂后得到目标产物(6.6g,98%),产率80%,淡黄色蜡状固体。
[0035]1h nmr(400mhz,cdcl3)δ4.27(m,2h),3.70
‑
3.65(m,60h),3.55(m,2h),3.38(s,3h),2.93(t,j=7.2hz,2h),2.79(t,j=7.2hz,2h),2.58(t,j=6.8hz,2h),2.08(s,3h),2.01(s,3h),1.97(s,3h),1.81
‑
1.75(m,2h),1.52(m,3h),1.38(m,3h),1.27
‑
1.23(m,12h),1.14(s,3h),1.07(s,3h),0.87
‑
0.84(m,12h);
13
c nmr(100mhz,cdcl3)δ172.2,170.9,149.5,140.6,126.7,125.0,123.0,117.4,,75.1,72.0,70.6,70.58
‑
70.33(m,多个碳),69.1,64.0,59.0,39.4,37.55
‑
37.29(m,多个碳),32.79
‑
32.69(m,4c),29.2,28.9,28.0,
24.816,24.805,24.5,22.8,22.7,21.1,20.6,19.77
‑
19.61(m,多个碳),1 13.0,12.1,11.8.
[0036]
实施例1:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0037]
反应式如下:
[0038][0039]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml 2wt.%的tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应8h,反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
1)所示的1,2
‑
二苯乙炔28.2mg,白色固体,收率93%,hplc检测纯度为99%。化合物式(ii
‑
1)的结构表征如下:
[0040]1h nmr(500mhz,cdcl3)δ7.59
‑
7.53(m,4h),7.43
‑
7.34(m,6h);
13
c nmr(126mhz,cdcl3)δ132.51,129.22,128.45,121.82,81.58,73.95;gc
‑
ms(ei):m/z202[m
].
[0041]
实施例2:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0042]
在空气气氛下,加入6mg(0.03mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应13h,反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
1)所示的1,2
‑
二苯乙炔11.5mg,白色固体,收率38%。
[0043]
实施例3:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0044]
在空气气氛下,加入18mg(0.09mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应6h,反应结束后向反应瓶加5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
1)所示的1,2
‑
二苯乙炔28.8mg,白色固体,收率95%,hplc检测纯度为99%。
[0045]
实施例4:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0046]
在空气气氛下,加入5mg(0.02mmol)催化剂cuso4·
5h2o到反应瓶中,然后加入30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应14h,反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在
真空减压下除去溶剂后得(ii
‑
1)所示的1,2
‑
二苯乙炔16.4mg,白色固体,收率54%,hplc检测纯度为95%。
[0047]
实施例5:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0048]
在空气气氛下,加入2.7mg(0.02mmol)催化剂cucl2到反应瓶中,然后加入30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应14h,反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
1)所示的1,2
‑
二苯乙炔19.7mg,白色固体,收率65%,hplc检测纯度为96%。
[0049]
实施例6:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0050]
在空气气氛下,加入1.98mg(0.02mmol)催化剂cucl到反应瓶中,然后加入30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应15h,反应结束后向反应瓶加5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
1)所示的1,2
‑
二苯乙炔9.7mg,白色固体,收率32%,hplc检测纯度为95%。
[0051]
实施例7:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0052]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml 2wt.%tpgs
‑
750
‑
m/h2o、12mg(0.3mmol)的naoh。常温下敞口搅拌反应15h,反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
1)所示的1,2
‑
二苯乙炔6.4mg,白色固体,收率21%,hplc检测纯度为95%。
[0053]
实施例8:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0054]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml 2wt.%tpgs
‑
750
‑
m/h2o、41.4mg(0.3mmol)的k2co3。常温下敞口搅拌反应8h,反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得痕量(ii
‑
1)所示的1,2
‑
二苯乙炔。
[0055]
实施例9:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0056]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入
30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml 2wt.%tpgs
‑
750
‑
m/h2o、30.3mg(0.3mmol)的et3n。常温下敞口搅拌反应12h,反应结束后向反应瓶加5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
1)所示的1,2
‑
二苯乙炔11.2mg,白色固体,收率37%,hplc检测纯度为96%。
[0057]
实施例10:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0058]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml 2wt.%tpgs
‑
750
‑
m/h2o、25.5mg(0.3mmol)的哌啶。常温下敞口搅拌反应8h,反应结束后向反应瓶加5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
1)所示的1,2
‑
二苯乙炔28.2mg,白色固体,收率93%,hplc检测纯度为99%。
[0059]
实施例11:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0060]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml 2wt.%tpgs
‑
750
‑
m/h2o、12.7mg(0.15mmol)的哌啶。常温下敞口搅拌反应13h,反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
1)所示的1,2
‑
二苯乙炔11.5mg,白色固体,收率38%,hplc检测纯度为96%。
[0061]
实施例12:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0062]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml 2wt.%tpgs
‑
750
‑
m/h2o、51mg(0.6mmol)的哌啶。常温下敞口搅拌反应7h,反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
1)所示的1,2
‑
二苯乙炔28.5mg,白色固体,收率92%,hplc检测纯度为99%。
[0063]
实施例13:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0064]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml trition x
‑
100水溶液、25.5mg(0.3mmol)的哌啶。常温下敞口搅拌反应15h,反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收
集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
1)所示的1,2
‑
二苯乙炔17.0mg,白色固体,收率56%,hplc检测纯度为97%。
[0065]
实施例14:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0066]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml sds(15mol%)的水溶液、25.5mg(0.3m mol)的哌啶。常温下敞口搅拌反应13h,反应结束后向反应瓶加5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
1)所示的1,2
‑
二苯乙炔10.0mg,白色固体,收率33%,hplc检测纯度为95%。
[0067]
实施例15:1
‑
甲基
‑4‑
[4
‑
(4
‑
甲基苯基)丁
‑
1,3
‑
二炔基]苯(ii
‑
2)的制备
[0068]
反应式如下:
[0069][0070]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入34.8mg(0.3mmol)对甲基苯乙炔(i
‑
2)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应7.5h,监测反应,原料(i
‑
2)基本反应完全。反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
2)所示的1
‑
甲基
‑4‑
[4
‑
(4
‑
甲基苯基)丁
‑
1,3
‑
二炔基]苯31.1mg,白色固体,收率90%,hplc检测纯度为98%。化合物式(ii
‑
2)的结构表征如下:
[0071]1h nmr(500mhz,cdcl3)δ7.44(d,j=8.1hz,4h),7.16(d,j=8.0hz,4h),2.39(s,6h);
13
c nmr(126mhz,cdcl3)δ139.50,132.41,129.23,118.83,81.57,73.49,21.62;gc
‑
ms(ei):m/z 230[m
].
[0072]
实施例16:1,4
‑
双(4
‑
戊基苯基)丁
‑
1,3
‑
二炔(ii
‑
3)的制备
[0073]
反应式如下:
[0074][0075]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入51.6mg(0.3mmol)对戊苯乙炔(i
‑
3)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。
常温下敞口搅拌反应8h,监测反应,原料(i
‑
3)基本反应完全。反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
3)所示的1,4
‑
双(4
‑
戊基苯基)丁
‑
1,3
‑
二炔47.2mg,白色固体,收率92%,hplc检测纯度为98%。化合物式(ii
‑
3)的结构表征如下:
[0076]1h nmr(500mhz,cdcl3)δ7.46(d,j=8.1hz,4h),7.17(d,j=8.0hz,4h),2.66
‑
2.60(m,4h),1.67
‑
1.60(m,4h),1.39
‑
1.30(m,8h),0.92(t,j=7.0hz,6h);
13
c nmr(126mhz,cdcl3)δ144.51,132.43,128.57,119.03,81.61,73.52,35.98,31.45,30.85,22.52,14.00;gc
‑
ms(ei):m/z 342[m
].
[0077]
实施例17:1,4
‑
双(4
‑
氟苯基)丁
‑
1,3
‑
二炔(ii
‑
4)的制备
[0078]
反应式如下:
[0079][0080]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入36.0mg(0.3mmol)对氟苯乙炔(i
‑
4)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应8h,监测反应,原料(i
‑
4)基本反应完全。反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
4)所示的1,4
‑
双(4
‑
氟苯基)丁
‑
1,3
‑
二炔33.2mg,白色固体,收率93%,hplc检测纯度为99%。化合物式(ii
‑
4)的结构表征如下:
[0081]1h nmr(500mhz,cdcl3)δ7.56
‑
7.50(m,4h),7.09
‑
7.02(m,4h);
13
c nmr(126mhz,cdcl3)δ163.10,134.56,117.86,115.93,80.46,73.57;gc
‑
ms(ei):m/z238[m
].
[0082]
实施例18:1,4
‑
双(3
‑
溴苯基)丁
‑
1,3
‑
二炔(ii
‑
5)的制备
[0083]
反应式如下:
[0084][0085]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入54.0mg(0.3mmol)间溴苯乙炔(i
‑
5)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应8h,监测反应,原料(i
‑
5)基本反应完全。反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂
后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
5)所示的1,4
‑
双(3
‑
溴苯基)丁
‑
1,3
‑
二炔47.3mg,白色固体,收率88%,hplc检测纯度为97%。化合物式(ii
‑
5)的结构表征如下:
[0086]1h nmr(500mhz,cdcl3)δ7.68(t,j=1.8hz,2h),7.55
‑
7.51(m,2h),7.47(d,j=7.8hz,2h),7.23(t,j=7.9hz,2h);
13
c nmr(126mhz,cdcl3)δ135.15,132.61,131.10,129.91,123.58,122.28,80.51,74.83;gc
‑
ms(ei):m/z 358[m
].
[0087]
实施例19:1,4
‑
双(2
‑
氯苯基)丁
‑
1,3
‑
二炔(ii
‑
6)的制备
[0088]
反应式如下:
[0089][0090]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入40.8mg(0.3mmol)邻氯苯乙炔(i
‑
6)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应8h,监测反应,原料(i
‑
6)基本反应完全。反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
6)所示的1,4
‑
双(2
‑
氯苯基)丁
‑
1,3
‑
二炔36.0mg,白色固体,收率89%,hplc检测纯度为96%。化合物式(ii
‑
6)的结构表征如下:
[0091]1h nmr(500mhz,cdcl3)δ7.59(dd,j=7.7,1.6hz,2h),7.44(dd,j=8.0,0.9hz,2h),7.33(td,j=7.8,1.7hz,2h),7.28
‑
7.25(m,2h);
13
c nmr(126mhz,cdcl3)δ137.00,134.40,130.30,129.48,126.58,121.85,79.44,78.41;gc
‑
ms(ei):m/z 270[m
].
[0092]
实施例20:1,4
‑
双(4
‑
甲氧苯基)丁
‑
1,3
‑
二炔(ii
‑
7)的制备
[0093]
反应式如下:
[0094][0095]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入39.6mg(0.3mmol)对甲氧基苯乙炔(i
‑
7)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应8h,监测反应,原料(i
‑
7)基本反应完全。反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
7)所示的1,4
‑
双(4
‑
甲氧苯基)丁
‑
1,3
‑
二炔(ii
‑
7)37.7mg,黄色固体,收率96%,hplc检测纯度为99%。化
合物式(ii
‑
7)的结构表征如下:
[0096]1h nmr(500mhz,cdcl3)δ7.45(d,j=8.7hz,4h),6.84(d,j=8.7hz,4h),3.80(s,6h);
13
c nmr(126mhz,cdcl3)δ160.24,134.02,114.13,113.92,81.24,72.97,55.30;gc
‑
ms(ei):m/z 262[m
].
[0097]
实施例21:1,4
‑
双(4
‑
乙氧苯基)丁
‑
1,3
‑
二炔(ii
‑
8)的制备
[0098]
反应式如下:
[0099][0100]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入43.8mg(0.3mmol)对乙氧基苯乙炔(i
‑
8)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应8h,监测反应,原料(i
‑
8)基本反应完全。反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
8)所示的1,4
‑
双(4
‑
乙氧苯基)丁
‑
1,3
‑
二炔(ii
‑
8)39.1mg,白色固体,收率90%,hplc检测纯度为97%。化合物式(ii
‑
8)的结构表征如下:
[0101]1h nmr(600mhz,cdcl3)δ7.47(d,j=8.8hz,4h),6.86(d,j=8.8hz,4h),4.09
‑
4.04(m,4h),1.44(t,j=7.0hz,6h);
13
c nmr(151mhz,cdcl3)δ159.64,134.04,114.62,113.76,81.30,72.90,63.59,14.73;gc
‑
ms(ei):m/z 290[m
].
[0102]
实施例22:1,4
‑
二(噻吩
‑3‑
基)丁
‑
1,3
‑
二炔(ii
‑
9)的制备
[0103]
反应式如下:
[0104][0105]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入32.4mg(0.3mmol)对3
‑
乙炔噻吩(i
‑
9)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应8h,监测反应,原料(i
‑
9)基本反应完全。反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
9)所示的1,4
‑
二(噻吩
‑3‑
基)丁
‑
1,3
‑
二炔(ii
‑
9)27.6mg,白色固体,收率86%,hplc检测纯度为96%。化合物式(ii
‑
9)的结构表征如下:
[0106]1h nmr(500mhz,cdcl3)δ7.61(dd,j=2.8,1.3hz,2h),7.30(dd,j=5.0,3.1hz,2h),7.19(dd,j=5.0,1.3hz,2h);
13
c nmr(126mhz,cdcl3)δ131.24,130.17,125.61,120.92,76.58,73.54;gc
‑
ms(ei):m/z 214[m
].
[0107]
实施例23:1,4
‑
双(炔丙基)丁
‑
1,3
‑
二炔(ii
‑
10)的制备
[0108]
反应式如下:
[0109][0110]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入34.8mg(0.3mmol)对炔丙基苯(i
‑
10)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应8h,监测反应,原料(i
‑
10)基本反应完全。反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
10)所示的1,4
‑
双(炔丙基)丁
‑
1,3
‑
二炔(ii
‑
10)32.8mg,白色固体,收率95%,hplc检测纯度为99%。化合物式(ii
‑
10)的结构表征如下:
[0111]1h nmr(500mhz,cdcl3)δ7.37(d,j=4.6hz,8h),7.31
‑
7.27(m,2h),3.73(s,4h);
13
c nmr(126mhz,cdcl3)δ135.56,128.62,127.95,126.86,75.56,67.27,25.61;gc
‑
ms(ei):m/z 230[m
].
[0112]
实施例24:1,4
‑
双(环丙基)丁
‑
1,3
‑
二炔(ii
‑
11)的制备
[0113]
反应式如下:
[0114][0115]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入19.8mg(0.3mmol)对环丙乙炔(i
‑
11)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应8h,监测反应,原料(i
‑
11)基本反应完全。反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
11)所示的1,4
‑
双(环丙基)丁
‑
1,3
‑
二炔(ii
‑
11)18.5mg,无色油状物,收率95%,hplc检测纯度为99%。化合物式(ii
‑
11)的结构表征如下:
[0116]1h nmr(600mhz,cdcl3)δ1.32
‑
1.28(m,2h),0.83
‑
0.79(m,4h),0.77
‑
0.73(m,4h);
13
c nmr(151mhz,cdcl3)δ80.05,60.85,8.70,0.02;gc
‑
ms(ei):m/z 130[m
].
[0117]
实施例25:1,4
‑
双(丁基)丁
‑
1,3
‑
二炔(ii
‑
12)的制备
[0118]
反应式如下:
[0119][0120]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入24.6mg(0.3mmol)对1
‑
己炔(i
‑
12)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。
常温下敞口搅拌反应8h,监测反应,原料(i
‑
12)基本反应完全。反应结束后向反应瓶加5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
12)所示的1,4
‑
双(丁基)丁
‑
1,3
‑
二炔(ii
‑
12)21.1mg,无色油状物,收率87%,hplc检测纯度为97%。化合物式(ii
‑
12)的结构表征如下:
[0121]1h nmr(500mhz,cdcl3)δ2.25(t,j=6.9hz,4h),1.53
‑
1.46(m,4h),1.44
‑
1.37(m,4h),0.90(t,j=7.3hz,6h);
13
c nmr(126mhz,cdcl3)δ77.38,65.25,30.38,21.89,18.85,13.48;gc
‑
ms(ei):m/z 162[m
].
[0122]
实施例26:1,4
‑
双(戊基)丁
‑
1,3
‑
二炔(ii
‑
13)的制备
[0123]
反应式如下:
[0124][0125]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入28.8mg(0.3mmol)对1
‑
庚炔(i
‑
13)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应8h,监测反应,原料(i
‑
13)基本反应完全。反应结束后向反应瓶加5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
13)所示的1,4
‑
双(戊基)丁
‑
1,3
‑
二炔(ii
‑
13)24.23mg,无色油状物,收率85%,hplc检测纯度为96%。化合物式(ii
‑
13)的结构表征如下:
[0126]1h nmr(500mhz,cdcl3)δ2.24(t,j=7.0hz,4h),1.58
‑
1.46(m,4h),1.39
‑
1.27(m,8h),0.89(t,j=7.1hz,6h);
13
c nmr(126mhz,cdcl3)δ77.42,65.30,31.00,28.06,22.16,19.15,13.87;gc
‑
ms(ei):m/z 190[m
].
[0127]
实施例27:1,4
‑
双(己基)丁
‑
1,3
‑
二炔(ii
‑
14)的制备
[0128]
反应式如下:
[0129][0130]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入33.0mg(0.3mmol)对1
‑
辛炔(i
‑
14)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应8h,监测反应,原料(i
‑
14)基本反应完全。反应结束后向反应瓶加5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
14)所示的1,4
‑
双(己基)丁
‑
1,3
‑
二炔(ii
‑
14)29.1mg,无色油状物,收率89%,hplc检测纯度为96%。化合物式(ii
‑
14)的结构表征如下:
[0131]1h nmr(500mhz,cdcl3)δ2.25(t,j=7.0hz,4h),1.56
‑
1.48(m,4h),1.42
‑
1.36(m,4h),1.34
‑
1.25(m,8h),0.89(t,j=6.9hz,6h);
13
c nmr(126mhz,cdcl3)δ77.49,65.30,31.32,28.55,28.35,22.53,19.22,14.02;gc
‑
ms(ei):m/z 218[m
].
[0132]
实施例28:1,4
‑
双(辛基)丁
‑
1,3
‑
二炔(ii
‑
15)的制备
[0133]
反应式如下:
[0134][0135]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入41.4mg(0.3mmol)对1
‑
奎炔(i
‑
15)、1ml 2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应8h,监测反应,原料(i
‑
15)基本反应完全。反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经柱层析分离(洗脱剂:石油醚:乙酸乙酯v/v=20:1),收集含目标化合物的洗脱液,在真空减压下除去溶剂后得(ii
‑
15)所示的1,4
‑
双(辛基)丁
‑
1,3
‑
二炔(ii
‑
15)36.2mg,无色油状物,收率88%,hplc检测纯度为96%。化合物式(ii
‑
15)的结构表征如下:
[0136]1h nmr(500mhz,cdcl3)δ2.25(t,j=6.9hz,4h),1.55
‑
1.49(m,4h),1.42
‑
1.25(m,20h),0.89(t,j=6.8hz,6h);
13
c nmr(126mhz,cdcl3)δ77.47,65.31,31.85,29.16,29.09,28.88,28.39,22.66,19.22,14.07;gc
‑
ms(ei):m/z 274[m
].
[0137]
实施例29:2,7
‑
二甲基
‑
3,5
‑
辛二炔
‑
2,7
‑
二醇(ii
‑
16)的制备
[0138]
反应式如下:
[0139][0140]
在空气气氛下,加入12mg(0.06mmol)催化剂cu(oac)2·
h2o到反应瓶中,然后加入25.2mg(0.3mmol)对2
‑
甲基
‑3‑
丁炔
‑2‑
醇(i
‑
16)、1ml2wt.%tpgs
‑
750
‑
m/h2o与25.5mg(0.3mmol)哌啶。常温下敞口搅拌反应8h,监测反应,原料(i
‑
16)基本反应完全。反应结束后向反应瓶加5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经薄层层析分离(石油醚:乙酸乙酯=20:1),经薄层层析分离(石油醚:乙酸乙酯=20:1),收集目标条带的硅胶并使用石油醚:乙酸乙酯v/v=20:1的溶剂对其进行洗脱,收集洗脱液并在真空减压下除去溶剂后制得(ii
‑
16)所示的1,4
‑
双(辛基)丁
‑
1,3
‑
二炔(ii
‑
16)20.4mg,无色油状物,收率82%,hplc检测纯度为96%。化合物式(ii
‑
16)的结构表征如下:
[0141]1h nmr(500mhz,dmso
‑
d6)δ5.56(s,2h),1.38(s,12h).
13
c nmr(126mhz,cdcl3)δ86.13,65.47,64.11,31.50;gc
‑
ms(ei):m/z 166[m
].
[0142]
对比例1:1,2
‑
二苯乙炔(ii
‑
1)的制备
[0143]
以苯乙炔为底物,在2%的tpgs750的参与下,在水相中不同催化剂催化端炔偶联反应,反应式式如下:
[0144][0145]
实验操作如下:
[0146]
在空气气氛下,加入催化剂(具体催化剂见表)到反应瓶中,然后加入30.9mg(0.3mmol)苯乙炔(i
‑
1)、1ml 2wt.%的tpgs
‑
750
‑
m/h2o与碱性物质。常温下敞口搅拌反应,反应结束后向反应瓶加入5ml(0.051mol)乙酸乙酯并搅拌5min。分离出有机相并真空减压下除去溶剂,经薄层层析分离(石油醚:乙酸乙酯=20:1),制得(ii
‑
1)所示的1,2
‑
二苯乙炔。
[0147]
反应结果如下表;
[0148][0149][0150]
a:以i
‑
1为基准的摩尔百分比;
[0151]
b:a为1h
‑
苯并三唑:
[0152][0153]
c:以i
‑
1为基准的摩尔当量;
[0154]
d:pd(η3‑
allyl)cl(pph3)
d
购买于萨恩化学技术(上海)有限公司,其结构式为:
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。