1.本发明涉及藏红曲有效成分检测技术领域,具体为一种藏红曲的质量控制方法和构建方法。
背景技术:
2.红曲为常用降脂药物,由红曲菌发酵而成,现临床使用的红曲类药物有2种,即中药“红曲”和藏药“藏红曲”。中药“红曲”,以梗米为基质,接种曲霉科真菌紫色红曲霉monascus purpureus went(cgmcc,no.0272菌株)发酵而成,又习称“红曲米”,临床使用有红曲饮片和其成药血脂康胶囊和脂必妥胶囊;藏药“藏红曲”(原名:青稞红曲),以我国藏区特产作物青稞(禾本科植物裸麦hordeum vulgare var.nudum hook.f.)的种仁作基质,接种曲霉科真菌红曲霉monascus pilosus(no.ywg
‑
1菌株)发酵而成,临床使用其饮片。
3.有研究报道,中药红曲降血脂的活性成分为洛伐他汀类成分,作为原料药材,其标准附于《中国药典》收载的“血脂康胶囊”条下,规定有“指纹图谱”、“洛伐他汀”的限量(不得少于0.22%)。藏红曲和中药红曲在“菌株、发酵基质、生产环境”等方面有一定差异,目前对藏红曲的质量标准却鲜有报道,鉴于藏红曲和中药红曲在“菌株、发酵基质、生产环境”等方面的差异,有必要对藏红曲的组成成分以及质量控制进行研究,建立反映出藏红曲的特色的标准,避免同质化。
技术实现要素:
4.本发明为了解决现有技术中存在的缺陷,提供一种藏红曲的质量控制方法和构建方法,所述方法通过uhplc检测,可在一次进样检测中同时确定出所述藏红曲成分的种类和含量,与现有中药红曲进行区分。
5.本发明首先提供一种藏红曲的质量控制方法,所述藏红曲的有效成分包括:核苷类化合物、红曲色素类化合物、洛伐他汀、桔霉素、莫拉可林类化合物;所述质量控制方法包括采用uhplc法检测所述藏红曲的有效成分。
6.优选的,所述uhplc法的色谱条件为:采用菲罗门c18色谱柱,柱温为30℃,流动相为甲醇
‑
水,进行梯度洗脱。
7.更为优选的,所述梯度洗脱过程中,流动相中甲醇浓度如下:
8.0~3min,6%甲醇;
9.3~7min,6%~16%甲醇;
10.7~10min,16%~16%甲醇,
11.10~15min,16%~60%甲醇,
12.15~25min,60%~85%甲醇,
13.25~32min,85%~85%甲醇,
14.32~34min,85%~100%甲醇,
15.34~38min,100%~100%甲醇,
16.38~40min,100%~6%甲醇,
17.40~44min,6%甲醇。
18.更为优选的,所述uhplc法的色谱条件还包括:检测波长为250nm。
19.更为优选的,所述流动相的流速为1ml/min。
20.更为优选的,所述uhplc法的色谱条件还包括:检测时进样量为10μl。
21.更为优选的,所述色谱柱的规格为:4.6mm
×
150mm,3.0μm。
22.本发明还提供一种由上述质量控制方法得到的指纹图谱,所述指纹图谱包括按照出峰时间顺序依次编号的21个色谱峰,其中,2、3、4号峰代表的成分均为核苷类物质,13、14、15号峰代表的成分均为红曲色素类物质,18号峰代表的成分为洛伐他汀,10号峰代表的成分为桔霉素,其他13个色谱峰代表的成分均为莫拉可林类化合物。
23.优选的,所述2号峰代表的成分为尿苷,所述3号峰代表的成分为鸟苷,所述4号峰代表的成分为腺苷,13号峰代表的成分的分子式为c
21
h
28
o5,14号峰代表的成分的分子式为c
21
h
26
o5,15号峰代表的成分的分子式为c
20
h
28
o4。
24.本发明还提供一种藏红曲质量控制构建方法,包括:
25.将藏红曲饮片碾碎处理后经渗透提取、减压浓缩,得藏红曲提取物;
26.将所述藏红曲提取物进行化学成分分段,得到分段提取物;
27.将所述藏红曲提取物和分段提取物采用上述uhplc色谱条件进行检测;
28.分析对比检测后得到的uhplc色谱图,确认对应所述藏红曲的特征峰信息。
29.优选的,所述渗透提取过程选用70%乙醇作为提取溶剂。
30.优选的,所述化学成分分段过程采用大孔树脂hpd
‑
100柱层析法。
31.更为优选的,所述化学成分分段过程具体为:
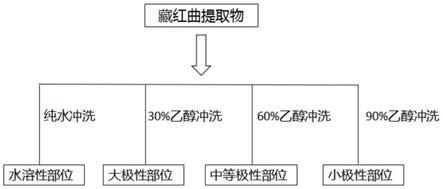
32.纯水冲洗所述藏红曲提取物,得到水溶性部位;
33.30%乙醇冲洗所述藏红曲提取物,得到大极性部位;
34.60%乙醇冲洗所述藏红曲提取物,得到中等极性部位;
35.90%乙醇冲洗所述藏红曲提取物,得到小极性部位;
36.优选的,莫拉可林类化合物主要分布在中等极性部位和小极性部位。
37.优选的,所述藏红曲中洛伐他汀的质量含量不得少于0.60%,所述洛伐他汀包括内酯式洛伐他汀和酸式洛伐他汀。
38.优选的,所述藏红曲中β
‑
葡聚糖的质量含量不得少于1.80%。
39.本发明的有益效果是:
40.1、本发明提供的藏红曲的质量控制方法中,针对藏红曲的成分特性,对液相色谱的条件进行了探索,可以一次性将藏红曲中的21种有效成分进行鉴定和检测,极大的提高了检测效率,较全面的反映了藏红曲的化学成分和品质特性,有利于藏红曲的质量控制,能够与现有中药红曲进行有效区分,避免同质化。
41.2、本发明限定uhplc法的色谱条件为采用菲罗门c18色谱柱,柱温为30℃,流动相为甲醇
‑
水,进行梯度洗脱。不同的色谱条件,高效液相色谱也会有显著不同,由于同时对所述藏红曲中的诸多有效成分进行检测鉴定,为了使各种有效成分进行有效分离而得到分离度较好的特征峰,本发明以指纹图谱呈现的色谱峰信息的丰富度以及主要有效成分洛伐他汀的提取率为考察指标,最终确定了色谱流动相洗脱条件,柱温,检测波长,在本发明条件
下,色谱峰从大极性到小极性的能较好的分离和呈现,使得单次检测即可得到分离度较好的峰,所述检测条件对藏红曲具有专属、稳定、实用的特性。
42.3、本发明的质量控制方法能够准确检测出藏红曲的有效成分,检测精密度高、重复性好、稳定性强、加样回收良好、耐用性较好。
43.4、本发明的藏红曲指纹图谱能较好的反映藏红曲产品的质量和稳定性,且可以鉴别出藏红曲饮片与临床常用的红曲类产品之间的差异;血脂康胶囊在成分组成上与藏红曲差异较大,均在0.85~0.88之间;其他中药红曲样品在成分上与藏红曲存在显著差异,相似度在0.33~0.81之间,反映出市场上不同厂家生产的红曲饮片的质量一致性较差。
44.5、本发明藏红曲质量控制构建方法,通过化学成分分段,使得藏红曲成分按照极性大小得到了较好的分段,为后续成分分离和药效研究提供了基础。
45.6、本发明的质量控制构建方法中,在所述色谱条件下,对20批次藏红曲样品的洛伐他汀含量测定,样品中洛伐他汀(内酯)和洛伐他汀(酸式)的总量在0.52%~0.87%,平均为0.75%,取均值下浮20%为0.60%,20批样品中18批合格,合格率为90%,据此确定洛伐他汀(内酯)和洛伐他汀(酸式)的总量限量为不得少于0.60%,能够对洛伐他汀进行质量控制。
46.7、本发明的质量控制构建方法中,在所述色谱条件下,对20批次藏红曲样品的β
‑
葡聚糖含量测定,β
‑
葡聚糖含量在1.75%
‑
3.17%,平均为2.31%,取均值下浮20%为1.8%,20批样品中17批合格,合格率为85.0%,据此确定含量限量为1.8%,能够对β
‑
葡聚糖进行质量控制。
附图说明
47.图1藏红曲提取物化学成分分段流程图
48.图2实施例1的uhplc
‑
uv色谱图(其中,s1:藏红曲总提取物,s2:纯水冲洗段,s3:30%乙醇冲洗段,s4:60%乙醇冲洗段,s5:90%乙醇冲洗段)
49.图3实施例1的uhplc
‑
ms色谱图(其中,1:藏红曲总提取物,2:纯水冲洗段,3:30%乙醇冲洗段,4:60%乙醇冲洗段,5:90%乙醇冲洗段)
50.图4实施例1藏红曲的对照指纹图谱中共有特征指纹峰
51.图5实施例3中28批藏红曲饮片的指纹图谱分析及全谱峰匹配图(其中,r:对照指纹图谱,s1~s28:藏红曲饮片样本)
52.图6实施例3中藏红曲对照指纹图谱(其中,1~10号峰为共有特征指纹峰,8号峰设为参照峰s)
53.图7血脂康胶囊样品与藏红曲对照指纹图谱的全谱峰匹配图(其中,r:藏红曲对照指纹图谱,s1~s12:血脂康胶囊样品)
54.图8中药红曲样品与藏红曲饮片对照指纹图谱的全谱峰匹配图(其中,r:藏红曲对照指纹图谱,s1~s10:中药红曲样品)
具体实施方式
55.为了使本领域的技术人员更好地理解发明的技术方案,下面结合具体实施方式对本发明作进一步的详细说明。
56.常规所说的红曲为曲霉科真菌红曲霉monascus purpureus went.的菌丝体寄生在粳米上而成的红曲米。分布于河北、江西、浙江、台湾、福建、广东等地。具有健脾消食,活血化瘀之功效。常用于饮食积滞,脘腹胀满,赤白下痢,产后恶露不尽,跌打损伤。
57.而藏红曲以西藏青稞为原料,通过青稞红曲发酵技术把红曲的抗氧化、降血脂等作用与青稞丰富的营养物质(如富含β
‑
葡聚糖、低脂肪、高纤维、高蛋白、高维生素)结合起来,使二者的有效成分得到充分利用,品质得到提升。
58.本发明为了建立反映出藏红曲特色的质量标准,避免与中药红曲同质化,提供一种藏红曲的质量控制方法和构建方法,所述方法通过uhplc(超高效液相色谱)检测,可在一次进样检测中同时确定出所述成分的种类和含量,与现有中药红曲进行区分。
59.为了实现上述目的,本发明首先提供一种藏红曲的质量控制方法,所述藏红曲的有效成分包括:核苷类化合物、红曲色素类化合物、洛伐他汀、桔霉素、莫拉可林类化合物;所述质量控制方法包括采用uhplc法检测所述藏红曲的有效成分。
60.本发明藏红曲中主要含有洛伐他汀类成分,同时还含有具有多种生物活性的β
‑
葡聚糖,这可能与藏红曲选择了适应高原环境的红曲菌株,以青稞为基质在高原(拉萨)发酵生产有关。由于同时对所述藏红曲中的诸多有效成分进行检测鉴定,为了使各种有效成分进行有效分离而得到分离度较好的特征峰,本发明对液相色谱的色谱条件进行了探索,使得单次检测即可得到分离度较好的峰。
61.不同的色谱条件,高效液相色谱也会有显著不同,本发明以指纹图谱呈现的色谱峰信息的丰富度以及主要有效成分洛伐他汀的提取率为考察指标,最终确定了色谱流动相洗脱条件,柱温,检测波长,在本条件下,色谱峰从大极性到小极性的能较好的分离和呈现,所述检测条件对藏红曲具有专属、稳定、实用的特性。在本发明中,所述uhplc法的色谱条件为:采用菲罗门c18色谱柱,柱温为30℃,流动相为甲醇
‑
水,进行梯度洗脱。
62.本发明所述梯度洗脱过程中,流动相中甲醇浓度如下:
63.0~3min,6%甲醇;
64.3~7min,6%~16%甲醇;
65.7~10min,16%~16%甲醇,
66.10~15min,16%~60%甲醇,
67.15~25min,60%~85%甲醇,
68.25~32min,85%~85%甲醇,
69.32~34min,85%~100%甲醇,
70.34~38min,100%~100%甲醇,
71.38~40min,100%~6%甲醇,
72.40~44min,6%甲醇。
73.所述uhplc法的色谱条件还包括:检测波长为250nm。
74.所述流动相的流速为1ml/min。
75.所述uhplc法的色谱条件还包括:检测时进样量为10μl。
76.所述色谱柱的规格为:4.6mm
×
150mm,3.0μm。
77.本发明还提供一种由上述质量控制方法得到的指纹图谱,所述指纹图谱包括按照出峰时间顺序依次编号的21个色谱峰,其中,2、3、4号峰代表的成分均为核苷类物质,13、
14、15号峰代表的成分均为红曲色素类物质,18号峰代表的成分为洛伐他汀,10号峰代表的成分为桔霉素,其他13个色谱峰代表的成分均为莫拉可林类化合物。进一步的,所述2号峰代表的成分为尿苷,所述3号峰代表的成分为鸟苷,所述4号峰代表的成分为腺苷,13号峰代表的成分的分子式为c
21
h
28
o5,14号峰代表的成分的分子式为c
21
h
26
o5,15号峰代表的成分的分子式为c
20
h
28
o4。
78.本发明还提供一种藏红曲质量控制构建方法,包括:
79.将藏红曲饮片碾碎处理后经渗透提取、减压浓缩,得藏红曲提取物;
80.将所述藏红曲提取物进行化学成分分段,得到分段提取物;
81.将所述藏红曲提取物和分段提取物采用上述uhplc色谱条件进行检测;
82.分析对比检测后得到的uhplc色谱图,确认对应所述藏红曲的特征峰信息。
83.在本发明技术方案中,由于藏红曲的有效成分较多,因此对提取溶剂的选择和提取方法需要特别考量。在本发明中,以指纹图谱呈现的色谱峰信息的丰富度以及主要有效成分洛伐他汀的提取率为考察指标,考察了制备方法的溶剂选择,最终确定了本发明中的质量控制构建方法,能比较全面的反映藏红曲中的从小极性到大极性的成分。
84.本发明中,所述渗透提取过程选用70%乙醇作为提取溶剂。具体为,将藏红曲饮片碾碎处理后加入其0.8倍质量的70%乙醇闷润12小时后层层加入渗漉筒,经70%乙醇渗漉提取得到渗漉提取液,收集渗漉液,边收集渗漉液边将渗漉液置于大型旋转蒸发仪进行减压浓缩。
85.本发明所述化学成分分段过程采用大孔树脂hpd
‑
100柱层析法,如图1所示,所述化学成分分段过程具体为:纯水冲洗所述藏红曲提取物,得到水溶性部位;30%乙醇冲洗所述藏红曲提取物,得到大极性部位;60%乙醇冲洗所述藏红曲提取物,得到中等极性部位;90%乙醇冲洗所述藏红曲提取物,得到小极性部位。莫拉可林类化合物主要分布在中等极性部位和小极性部位。
86.本发明藏红曲中洛伐他汀的含量为洛伐他汀(内酯)和洛伐他汀(酸式)的总量不得少于0.60%。
87.本发明藏红曲中β
‑
葡聚糖的含量不得少于1.80%。
88.上述为本发明的详细阐述,下面为本发明实施例,本发明实施例中藏红曲均来自西藏月王生物科技有限公司。
89.实施例1藏红曲样品质量控制构建方法
90.试验仪器:kq
‑
300de型数控超声波清洗器(功率300w,频率可调,昆山市超声仪器有限公司);milli
‑
q超纯水仪(美国密理博公司);电热恒温鼓风干燥箱(上海新苗医疗器械制造有限公司);酒精计;bt25s型电子分析天平(北京赛多利斯仪器系统有限公司);r
‑
210型旋转薄膜转蒸发仪(瑞士buchi公司);lc
‑
20atxr岛津液相色谱仪(日本岛津仪器);ab sicex tof5600 质谱仪(美国爱博才思仪器公司);一次性使用无菌注射器(带针)圣光医用制品股份有限公司;一次性0.22、0.45μm滤膜广州市文睿科学仪器有限公司;ymc c18色谱柱(日本ymc科技);
91.质谱条件:电喷雾电离,正、负2种离子模式检测,氮气作为鞘气、辅助气和吹扫气。正离子模式下:喷雾电压3.5kv,鞘气3.5mpa,辅助气785kpa,吹扫气0pa;毛细管温度270℃,毛细管电压为10v,透镜电压为80v。负离子模式下:喷雾电压5kv,鞘气2.7mpa,辅助气
785kpa,吹扫气0pa;毛细管温度270℃,毛细管电压为
‑
10v,透镜电压为
‑
100v。全扫描质量范围:m/z 100~1500。二级质谱数据依赖模式采集,全扫描图谱中响应度强度最高的3个峰用于二级质谱分析,高纯氦气用作碰撞气,碰撞能量为35ev。
92.试验试剂:样品制备用乙醇为分析纯,购自西陇化工股份有限公司;液相色谱所用甲醇购为色谱纯(批号:654655
‑
5456,美国天地公司),水为实验室密理博纯水仪产超纯水。
93.本实施例中的藏红曲质量控制构建方法采用如下步骤:
94.1、将藏红曲饮片碾碎处理后加入其0.8倍质量的70%乙醇闷润12小时后层层加入渗漉筒,经70%乙醇渗漉提取得到渗漉提取液,收集渗漉液,边收集渗漉液边将渗漉液置于大型旋转蒸发仪进行减压浓缩,得藏红曲提取物。
95.2、采用大孔树脂hpd
‑
100柱层析法将所述藏红曲提取物进行化学成分分段,具体为:用纯水冲洗所述藏红曲提取物,得到水溶性部位;用30%乙醇冲洗所述藏红曲提取物,得到大极性部位;用60%乙醇冲洗所述藏红曲提取物,得到中等极性部位;用90%乙醇冲洗所述藏红曲提取物,得到小极性部位;得到各分段提取物。
96.3、将所述藏红曲提取物和分段提取物采用uhplc进行检测;分析对比检测后得到的uhplc色谱图,确认对应所述藏红曲的特征峰信息。
97.所述uhplc的色谱条件为:色谱柱为菲罗门c18色谱柱(luna,4.6mm
×
150mm,3.0μm),柱温30℃,流速1ml/min,检测波长为250nm,进样量10μl。流动相为甲醇
‑
水,梯度洗脱程序:
98.0~3min,6%甲醇;
99.3~7min,6%~16%甲醇;
100.7~10min,16%~16%甲醇,
101.10~15min,16%~60%甲醇,
102.15~25min,60%~85%甲醇,
103.25~32min,85%~85%甲醇,
104.32~34min,85%~100%甲醇,
105.34~38min,100%~100%甲醇,
106.38~40min,100%~6%甲醇,
107.40~44min,6%甲醇。
108.所得分析结果见图2(,s1:藏红曲总提取物,s2:纯水冲洗段,s3:30%乙醇冲洗段,s4:60%乙醇冲洗段,s5:90%乙醇冲洗段))和图3(1:藏红曲总提取物,2:纯水冲洗段,3:30%乙醇冲洗段,4:60%乙醇冲洗段,5:90%乙醇冲洗段),可以看出,各段成分差别明显,藏红曲成分按照极性大小得到了较好的分段。
109.通过色谱峰的紫外以及质谱碎片信息进行数据库搜索和文献比对藏红曲对照指纹图谱中21个共有特征峰进行初步鉴定,共鉴定出成分,各峰编号见图4,各化合物鉴定结果质谱相关信息表见表1。在鉴定出的成分中,2、3、4号峰代表的成分均为核苷类物质,13、14、15号峰代表的成分均为红曲色素类物质,18号峰为藏红曲饮片主要活性成分洛伐他汀(monacolink),10号峰为主要毒性成分桔霉素(cirinin);其他13个色谱峰代表的成分均为莫拉可林(monacolins)类化合物,结果表明藏红曲饮片中含有多种莫拉可林类(他汀类)成分,且其中洛伐他汀,即monacolin k为这类成分中含量较高的代表性成分。
110.表1 藏红曲提取物中成分的质谱鉴定信息表
111.112.[0113][0114]
实施例2藏红曲样品hplc指纹图谱方法学考察
[0115]
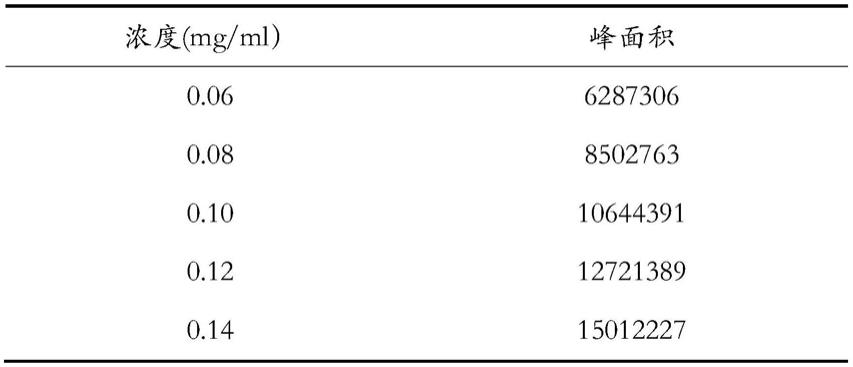
精密度试验:按实施例1的质量控制构建条件,分别精密吸取洛伐他汀(内酯)对照品溶液(0.1054mg/ml)和洛伐他汀(酸式)对照品溶液(0.1243mg/ml)10μl,注入液相色谱仪,测定,每个对照品溶液重复进样6次,记录峰面积。精密度试验结果见表2。
[0116]
表2 精密度试验结果
[0117][0118]
结果表明:上述测得洛伐他汀(内酯)和洛伐他汀(酸式)的对照品峰面积rsd分别为0.93%和0.19%,均小于2.0%,表明系统精密度良好。
[0119]
重复性试验:取不同批号的藏红曲样品约0.5g,精密称定6份,置具塞锥形瓶中,精密加入30%甲醇25ml,密塞,称定重量,超声处理(功率140w,频率42khz)1.5小时,放冷,再称定重量,用甲醇补足减失的重量,高速离心提取液,取上清液经0.45μml滤头滤过,取续滤液即得,按实施例1的质量控制构建条件,进样,测定,重复性考察结果见表3
‑
表5。
[0120]
表3 重复性试验结果(样品批号20170317)
[0121][0122][0123]
表4 重复性试验结果(样品批号20170612)
[0124][0125]
表5 重复性试验结果(样品批号20181101)
[0126][0127]
结果表明,三批样品得平均含量分别为0.8208%、0.7119%和0.8680%;rsd分别为1.90%、1.86%和0.46%,表明方法重复性良好。
[0128]
稳定性试验:取不同批号的藏红曲样品,按实施例1的质量控制构建条件制备,间隔进样,每次进样间隔两小时,分别在0h、2h、4h、6h、8h及12h进行测定,记录峰面积,稳定性考察结果见表6
‑
表8。
[0129]
表6 稳定性考察结果(样品批号20170317)
[0130][0131]
表7 稳定性考察结果(样品批号20170810)
[0132][0133]
表8 稳定性考察结果(样品批号20180605)
[0134][0135]
结果表明,上述三批样品测得的洛伐他汀(内酯)和洛伐他汀(酸式)峰面积rsd分别为0.17%和0.12%、0.01%和0.69%、0.07%和0.27%。表明在本发明条件下,供试品溶液在12小时内稳定。
[0136]
加样回收试验:取不同批号的藏红曲样品各6份,每份约0.5g,精密称定,置具塞锥形瓶中,分别精密加入浓度为0.0635mg/ml的洛伐他汀(内酯)对照品溶液25ml和浓度为0.0434mg/ml的洛伐他汀(酸式)对照品溶液25ml,按按实施例1的质量控制构建条件测定,计算回收率。结果见表9
‑
表11。
[0137]
表9 加样回收试验(样品批号20170317)
[0138][0139]
表10 加样回收试验(样品批号20170612)
[0140]
[0141]
表11 样回收试验(样品批号20181101)
[0142][0143][0144]
结果表明,三批样品洛伐他汀的平均回收率分别为103.44%、101.86%和101.87%,rsd(n=6)分别为1.52%、1.83%和1.28%。样品回收率在95%
‑
105%之间,加样回收良好。
[0145]
耐用性试验:取不同批号的藏红曲样品按实施例1的质量控制构建条件制备,分别使用agilent 1260 infinity hplc仪系统(美国安捷伦)和essentia
‑
16 hplc仪系统(日本岛津),按实施例1的色谱条件,进样测定。结果见表12
‑
14。
[0146]
表12 耐用性试验结果(样品批号20170317)
[0147][0148]
表13 耐用性试验结果(样品批号20170612)
[0149][0150]
表14 耐用性试验结果(样品批号20181101)
[0151]
[0152][0153]
结果表明,三批样品采用不同仪器测定的含量rsd分别为1.68%、0.94%和0.54%,表明方法的耐用性较好。
[0154]
实施例3藏红曲饮片hplc指纹图谱分析
[0155]
检测条件同实施例1,取14批藏红曲饮片样品,每批样品取2个样本,对28个样本的色谱数据中保留时间在2~38min内峰高大于1000的色谱峰自动积分,以批号为20080201的一个样本色谱图为参照图谱,中位数生成法,选取时间宽度为0.1min,将28个样本色谱数据导入中药色谱指纹图谱相似度评价系统(2012版),通过比较选择6个共有特征成分峰作为marker峰进行多点校正,进行全谱峰匹配得到峰匹配指纹图谱(图5,r:对照指纹图谱;s1~s28:藏红曲饮片样本),软件生成对照指纹图谱(图6,1~10号峰为共有特征指纹峰,8号峰设为参照峰s),通过软件相似度分析,得到各批次样品的相似度数据,结果见表15。
[0156]
表15 28批藏红曲饮片样本与对照图谱相似度分析结果
[0157]
[0158][0159]
结果显示,本发明建立的藏红曲饮片对照指纹图谱与28个样本之间的相似度均在0.95以上,表明所用各批次藏红曲饮片样品质量稳定性很好,批次间差异较小。
[0160]
藏红曲与中成药血脂康胶囊的指纹图谱对比分析
[0161]
检测条件同实施例1,采用6批血脂康胶囊,每批样品取2个样本,药品来源见表16,将所得色谱数据及藏红曲对照指纹图谱导入中药色谱指纹图谱相似度评价系统(2012版),计算相似度,结果见图7(r为藏红曲对照指纹图谱,s1~s12为血脂康胶囊样品)和表17。
[0162]
表16 血脂康样品批号与来源
[0163]
药品样品批号来源血脂康181205北大维信血脂康171223北大维信血脂康181221北大维信血脂康190312北大维信血脂康190406北大维信血脂康190411北大维信
[0164]
表17 血脂康胶囊与藏红曲对照指纹图谱相似度分析结果
[0165][0166]
结果表明,血脂康胶囊各样本与藏红曲对照指纹图谱的相似度均在0.85~0.88之间,表明血脂康胶囊与藏红曲饮片在成分组成上存在一定差异。
[0167]
藏红曲饮片与其他中药红曲样品的指纹图谱对比分析
[0168]
检测条件同实施例1,检测中药红曲样品5批共计10个样本(每批样品取2个样本),药品来源见表18,将所得色谱数据及藏红曲饮片对照指纹图谱导入中药色谱指纹图谱相似度评价系统(2012版),计算相似度,结果见图8(r为藏红曲饮片对照指纹图谱;s1~s10为中药红曲样品)和表19。
[0169]
表18 中药红曲样品批号与来源
[0170]
药品样品批号来源药品22中药红曲饮片181122浙江桐君堂中药23中药红曲饮片160407宝鸡汉方国药24中药红曲饮片1812152北京盛世龙药业25中药红曲饮片041812051北京万泰利克药业26中药红曲饮片1807249四川千方中药
[0171]
表19 中药红曲样品与藏红曲饮片对照指纹图谱相似度分析结果
[0172][0173]
结果表明,上述5个厂家的中药红曲样本与藏红曲对照指纹图谱的相似度最低为0.33,最高为0.81,表明中药红曲样本在成分组成上与藏红曲存在较大差异,且各批次样品间质量稳定性差。
[0174]
以上对藏红曲与其他中药红曲类各样本的指纹图谱分析表明,本发明的藏红曲对照指纹图谱能较好的反映藏红曲饮片产品的质量和稳定性,且可以鉴别出藏红曲饮片与临床常用的红曲类产品之间的差异;血脂康胶囊在成分组成上与藏红曲差异较大,均在0.85~0.88之间;其他中药红曲样品在成分上与藏红曲存在显著差异,相似度在0.33~0.81之间,反映出市场上不同厂家生产的红曲饮片的质量一致性较差。
[0175]
实施例4藏红曲的质量控制标准
[0176]
洛伐他汀含量测定
[0177]
取20个批次的藏红曲样品,分别取样品粉末(过三号筛)约0.5g,精密称定,置50ml具塞容量瓶中,加75%乙醇30ml,密塞,超声处理(功率250w,频率28khz)50分钟,放冷,加75%乙醇至接近刻度,再超声处理10分钟,放冷,用75%乙醇定容至刻度,摇匀,取上清液经0.45μm微孔滤膜滤过,取续滤液,即得各供试品溶液。按实施例1所述色谱条件测定,记录峰面积,计算含量,结果见表20。
[0178]
表20 20批次藏红曲供试样品的洛伐他汀含量测定结果
[0179][0180]
结果表明,20批藏红曲样品中洛伐他汀(内酯)和洛伐他汀(酸式)的总量在0.52%~0.87%,平均为0.75%,取均值下浮20%为0.60%,20批样品中18批合格,合格率为90%,据此确定洛伐他汀(内酯)和洛伐他汀(酸式)的总量限量为不得少于0.60%。
[0181]
β
‑
葡聚糖含量测定(紫外
‑
可见分光光度法(通则0401)测定)
[0182]
1、相关试剂和溶液的配制
[0183]
(1)50u/ml地衣聚糖酶溶液的配制
[0184]
取lml地衣聚糖酶溶液(1000u/ml),加磷酸钠缓冲液(20mmol/l,ph6.5)稀释定容至20.0ml,将酶液等分成4份,各5ml,置于聚丙烯塑料瓶中,在
‑
20℃下冷冻保存,备用。[注:地衣聚糖酶活力单位定义为:在ph6.5、40℃条件下,每分钟从大麦β
‑
葡聚糖(10mg/ml)中释放出相当于1μmol/l的葡萄糖还原糖当量所需的酶量,即为1u。]
[0185]
(2)2u/mlβ
‑
葡萄糖苷酶溶液的配制
[0186]
取1mlβ
‑
葡萄糖苷酶溶液(40u/ml),加乙酸钠缓冲液(50mmol/l,ph=4.0)稀释定容至20ml,将酶液等分成4份,各5ml,置于聚丙烯塑料瓶中,在
‑
20℃下冷冻保存,备用。[注:β
‑
葡萄糖苷酶活力单位定义为:在ph=4.0、40℃条件下,每分钟从对
‑
硝基苯基
‑
β
‑
葡萄糖
苷(10mmol)中释放出相当于lμmol/l的对
‑
硝基苯酚当量所需的酶量,即为1u。]
[0187]
(3)葡萄糖氧化酶
‑
过氧化物酶
‑
缓冲液混合溶液的配制
[0188]
内含葡萄糖氧化酶(>12000u/l)、过氧化物酶(>650u/l)、4
‑
氨基安替吡啉(0.4mmol/l)。[注:葡萄糖氧化酶活力单位定义为:在ph5.1、35℃条件下,每分钟氧化1mmol的葡萄糖所需的酶量,即为1u;过氧化物酶活力单位定义为:在ph6.0、20℃条件下,20秒内使焦性没食子酸生成1.0mg红棓酚所需的酶量,即为1u]
[0189]
缓冲液的配制:取13.6g kh2po4、4.2g naoh和3.0g 4
‑
羟基苯甲酸,加水溶解,定容至100ml,调节ph至7.4,即得。
[0190]
葡萄糖氧化酶
‑
过氧化物酶
‑
缓冲液混合物的配制:取50ml缓冲液稀释至1000ml;用该稀释缓冲液溶解葡萄糖氧化酶
‑
过氧化物酶混合试剂,使之达到所需浓度,即得。
[0191]
(4)50%乙醇溶液配制:取无水乙醇50ml,用水稀释至100ml。
[0192]
(5)20mmol/l、ph6.5磷酸钠缓冲液的配制
[0193]
取3.12g nah2p04·
2h20,加水900ml溶解,定容至l000ml,调节ph至6.5,即得。
[0194]
(6)乙酸钠缓冲液的配制
[0195]
200mmol/l、ph4.0乙酸钠缓冲液:取7.6ml冰醋酸,加水稀释至900ml,加4.8g三水乙酸钠,振摇使溶解;调节ph至4.0,定容至l000ml。
[0196]
50mmol/l、ph4.0乙酸钠缓冲液:取250ml 200mmol/l、ph4.0乙酸钠缓冲液稀释至1000ml。
[0197]
2、d
‑
葡萄糖标准储备液的制备
[0198]
取对照品粉末(纯度大于99%)于100℃下常压干燥2小时;置于干燥器中冷却,室温下密闭保存。取干燥后的葡萄糖对照品约0.1000g,精密称定,用50mmol/l、ph4.0乙酸钠缓冲液,振摇使溶解,并定容至100ml,即得。
[0199]
注:上述步骤中,涉及酶制剂均可由megazyme混联β
‑
d葡聚糖测定试剂盒提供,megazyme是爱尔兰megazyme公司产品的商品名。
[0200]
3、供试品溶液的制备与测定法
[0201]
(1)取样取本品约0.1g,精密称定,置于试管底部,酶解前处理向待测样品试管中添加0.5ml 50%乙醇溶液,于漩涡混合仪上振荡分散;加4.0ml磷酸钠缓冲液,超声处理5分钟。将试管置沸水浴中保持1分钟;取出试管,在漩涡混合仪上剧烈振荡数秒;沸水浴中继续保持2分钟,再振荡处理同前。
[0202]
(2)酶解反应将试管置50℃水浴中保温5分钟,加0.2ml地衣聚糖酶溶液,剧烈振荡数秒;加盖试管塞,50℃水浴继续保温60min;期间将试管取出,振荡处理3次
‑
4次。取出试管,加入5ml 200mmol/l乙酸钠缓冲液,混匀,室温下冷却5
‑
10分钟;离心(1000rad/min,10min),取上清液,备用。分别准确移取0.3ml上清液至3支试管的底部,向其中2支试管中加入0.3mlβ
‑
葡萄糖苷酶溶液;另一只试管中加入0.3ml 50mmol/l乙酸钠缓冲液作为反应空白;将各试管在50℃下保温10分钟。
[0203]
(3)葡萄糖标准工作液:平行移取d
‑
葡萄糖标准储备液0.15ml于2支试管中,分别加入0.15ml 50mmol/l乙酸钠缓冲液,即得。
[0204]
(4)试剂空白:移取0.3ml 50mmol/l乙酸钠缓冲液至试管中,即得。
[0205]
(5)显示反应分别移取9.0ml葡萄糖氧化酶
‑
过氧化物酶
‑
缓冲液混合物至试管(包
括2个测试样、1个反应空白、3个d
‑
葡萄糖标准工作溶液、1个试剂空白),在50℃下反应20分钟;取出试管,冷却至室温。
[0206]
(6)含量测定以试剂空白调零,其他试液于510nm处测定吸光值,计算结果。(如果试样中β
‑
葡聚糖的浓度大于10%,将产生比100μg葡萄糖标准工作溶液高的吸光度。此时需在酶解反应振荡处理后,取适量50mmol/l乙酸钠缓冲液稀释上清液,然后继续后面的步骤,结果计算时应把稀释因子考虑在内。)
[0207]
结果计算:
[0208][0209]
a1——样品测定液吸光值;
[0210]
a0——反应空白吸光值;
[0211]
a——d
‑
葡萄糖标准工作液吸光值;
[0212]
c——d
‑
葡萄糖标准储备液浓度,单位微克每毫升(μg/ml);
[0213]
——体积校正因子(从9.7ml移取0.15ml用于分析);
[0214]
m——样品质量,单位为克(g);
[0215]
0.9——d
‑
葡萄糖转化为β
‑
葡聚糖的脱水转换因子。
[0216]
4、样品测定
[0217]
20批藏红曲饮片样品测定结果见表21。
[0218]
表21 藏红曲样品中β
‑
葡聚糖含量测定结果
[0219]
[0220]
[0221][0222]
结果表明,20批藏红曲饮片样品的β
‑
葡聚糖含量在1.75%
‑
3.17%,平均为2.31%,取均值下浮20%为1.8%,20批样品中17批合格,合格率为85.0%,据此确定含量限量为1.8%。
[0223]
以上仅是本发明的优选实施方式,应当指出的是,上述优选实施方式不应视为对本发明的限制,本发明的保护范围应当以权利要求所限定的范围为准。对于本技术领域的普通技术人员来说,在不脱离本发明的精神和范围内,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。