预测前列腺癌的方法及其用途
1.相关申请的交叉引用
2.本技术要求2020年2月18日提交的美国临时申请第62/978,184号和2019年3月15日提交的美国临时申请第62/819,325号的权益,它们的全部内容通过引用并入本文。
技术领域
3.本发明涉及用于诊断、预后、监测和治疗前列腺癌患者的组合物和方法。特别地,本发明涉及小非编码rna(sncrna)例如mirna和snorna作为表达特征用于鉴定具有临床意义的前列腺癌的用途。
背景技术:
4.当前的前列腺癌筛查方法包括直肠指检,然后是前列腺特异性抗原(psa)测试。前者是侵入性的,后者需要从受试者身上抽取血样。
5.对可疑dre和/或psa水平升高的患者进行系统的12针核心活检或磁共振成像(mri)引导的靶向针活检。这种标准的诊断策略是侵入性的、不精确的,并且与高成本的发病率、最显著的细菌感染相关。
6.psa测试有明显的缺点。除了表明前列腺癌,升高的psa水平还可能表明尿路感染或前列腺炎(炎症或前列腺或良性前列腺增生或bph)。该测试过度诊断了前列腺癌,许多男性不必要地接受了核心针活检。在活检期间收集的前列腺组织然后由病理学家检查并分配gleason分数,以评估疾病的等级。gleason分数是两个数字的和:(1)病理学家根据病理学家对最常见病理学中肿瘤分级的确定分配的初级等级,(2)基于在其次最突出的病理学中肿瘤等级确定的二级等级。对于每个区域,根据肿瘤出现的侵袭程度分配一到五的分数,并将这两个数字加在一起以提供最终的gleason分数。细胞看起来接近正常的肿瘤被指定为低gleason分数(六分或以下,报告为gleason 3 3),而细胞与正常前列腺细胞明显不同的肿瘤被指定为较高的gleason分数(七或以上)。基于低gleason分数的低等级肿瘤不太可能具有侵袭性;而具有高gleason分数的肿瘤更有可能具有侵袭性和转移性。自实施以来,gleason评分系统的某些方面一直存在问题——最值得注意的是,gleason 3 4和gleason 4 3的肿瘤都被报告为gleason 7,即使这些组的临床结果明显不同。最近对gleason分数进行了改进,称为等级分组,已被采用来消除这个问题(等级组1(gg1)包括gleason 3 3;gg2
‑
gleason 3 4;gg3
‑
gleason 4 3;gg4
‑
gleason 4 4和gg5
‑
gleason 5 4或更高。评分系统的这一变化简化了前列腺癌组织病理学的报告,并消除了与“gleason 7”肿瘤相关的歧义,使分类结果更加直接。
7.大约50
‑
70%的基于“升高”的psa(>3ng/ml)被推荐进行核心针活检的患者的活检结果为阴性,而14%psa<3ng/ml的男性患有前列腺癌,但常规上并不因其psa水平低而进行活检。psa筛查和核心针活检的结合既是侵入性的,又具有较差的性能特征,这使得医生和患者没有可靠的措施来选择治疗选项。结果是许多男性不必要地选择了临床干预,通常是前列腺切除术。它还阻碍了新预后工具的开发,因为gleason分数“黄金标准”本身并不是前
列腺肿瘤进展的可靠指标。
8.这个问题已经被认识到至少30年。这在今天仍然是一个主要问题。在此年间,有许多尝试开发侵袭性疾病的预后标志物,包括倍性、核形态和核基质结构、基于微阵列的转录组分析、dna甲基化状态和基因融合检测,如tmprss2:ets家族融合。这些方法中没有一个被证明比作为前列腺肿瘤进展指标的gleason分数显著更好。此外,他们没有充分确定癌症分期或等级。
9.已经开发出旨在使用mrna表达谱区分癌症状态的测试。然而,每一个都表现出显著的缺点。首先,除了少数例外,这些检测使用来自根治性前列腺切除术标本的肿瘤材料,因此最多可以预测早期肿瘤复发。虽然可能有助于做出与持续临床决策相关的术后临床决策,但它们无助于区分手术前的前列腺癌等级。其次,许多这些基因组方法都集中在与前列腺癌进展有关的特定途径上,包括雄激素受体(ar)调节的基因表达、上皮
‑
基质相互作用和细胞周期。这些测定假设所有前列腺肿瘤都沿着共同的途径发展。其他商业上可用的生物标志物检测使用由一小部分基因的实时pcr生成的mrna表达谱。
10.迄今为止,很少有关于前列腺癌中sncrna的全基因组转录组研究。在包含在sanger mirbase v.10.1(wellcome sanger institute)中编目为723人mirna的affymetrixv.2微阵列上,使用illumina/solexa深度测序和微阵列分析,一项研究比较了(i)新鲜冷冻的前列腺癌根治术的样品和(ii)同一患者邻近的正常组织中的mirna和snorna特征。该研究为比较在前列腺癌和肿瘤周围良性组织中表达的sncrna的补充提供了一个有价值的数据集,但对于在临床干预前对肿瘤进展具有预后和/或预测性的一组sncrna的合理设计没有用。它作为一般筛查技术也有缺陷,因为该技术需要微解剖的快速冷冻材料,只能在手术后使用,因此不能用于诊断。
11.因此,需要一种预测筛查和分类前列腺癌的改进方法。本公开涉及一种用于筛查前列腺癌存在与否的非侵入性(通过消除或减少不必要的核心针活检)方法,该方法具有高度敏感性和特异性。
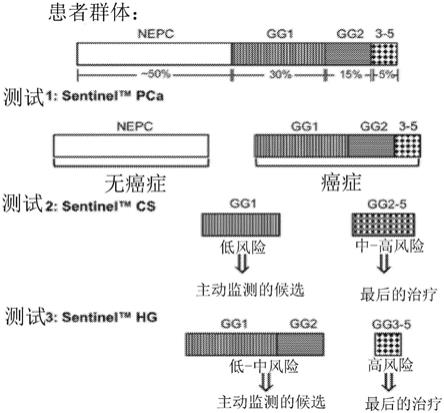
12.该方法还提供了用于疾病管理的平台,该平台可用于疾病的诊断、分类、预后,以及进展和治疗的监测。公开的方法基于结合sentinel
tm
pca、sentinel
tm
cs和sentinel
tm
hg测试对从尿外来体中分离的大量至少200个小非编码rna(sncrna)进行询问。sentinel
tm
pca、sentinel
tm
cs和sentinel
tm
hg测试基于snrna序列的算法分析和比较,该序列从大型目标人群编目,该人群对于sentinel
tm
pca测试,无前列腺癌证据(nepc)或具有前列腺癌(gg1
‑
gg5);对于sentinel
tm
cs测试,具有低等级癌症(gg1)与中和高等级癌症(gg2
‑
gg5)比较;并且对于sentinel
tm
hg测试,具有低和有利的中等级癌症(gg1 gg2)与不利的中和高等级(gg3
‑
gg5)癌症。可在单个尿样上进行的三个sentinel
tm
测试用于依次确定患者是否患有前列腺癌,以及前列腺癌患者是否患有可以在主动监督协议上监测的低或有利的中等级疾病或需要立即治疗的高级别疾病。
技术实现要素:
13.一方面,本公开提供了一种筛查受试者的前列腺癌的方法,包括:(i)从受试者获得生物样品,(ii)检测来自所述生物样品的小非编码rna(sncrna)特征集合的聚集表达谱,其中sncrna的所述集合包含seq id no:1
‑
280,(iii)通过在来自无前列腺癌证据(nepc)或
前列腺癌的风险的受试者的可能性,和(iv)通过施用一种或多种化学治疗剂、激素、免疫疗法、放射、冷冻疗法、手术或它们的组合,治疗预测具有发展侵袭性前列腺癌的高风险的受试者。
19.在另一方面,本公开提供了一种系统,其用于确定患者是否患有癌症或患有癌症并将患有癌症的受试者分类为(i)惰性(低等级,gg1)、(ii)中或高等级(gg2
‑
gg5)、(iii)低/中风险(gg1
‑
gg2)或(iv)侵袭性(高等级,gg3
‑
gg5)前列腺癌,所述系统包括至少三个处理器,其配置为(a)询问sncrna序列的信息序列,(b)确定和比较sentinel分数以确定受试者是否患有前列腺癌或没有前列腺癌,并对前列腺癌分期组进行分类。
附图说明
20.本专利或申请文件含有至少一幅彩色附图。在请求并支付必要的费用后,官方将会提供带有一幅或多幅彩色附图的本专利或专利申请公开物的副本。
21.图1:外来体是在早期内体中形成并从细胞中释放的小的细胞外膜结合囊泡。外来体包含蛋白质、mrna和反映细胞生物学的sncrna阵列[mirna和c/d盒和h/aca盒小核仁rna(snorna)]。
[0022]
图2:使用商业试剂盒,从患者收集无细胞尿液。捕获尿液中的外来体,并从外来体中提取rna。使用qubit分析(thermofisher)测量rna产量,使用agilent 2100生物分析仪评估样品质量。使用专门为mir scientific sentinel
tm
pca、sentinel
tm
cs或sentinel
tm
hg测试设计的定制openarray
tm
(thermo fisher)板对所得sncrna水平进行询问。询问是同时分析生物样品中大量序列的通用技术术语。然后对snorna和microrna(统称为sncrna)的扩增曲线的所得读数进行分析并用于诊断患者,并在出现癌症时对疾病进行分类,并相应地监测治疗。
[0023]
图3说明了使用多于单个实体建立无偏统计方法的复杂性,以识别重要的相互作用,以识别sncrna的个体和组合,并关联等级分组或前列腺癌表型。(参见[00048]
‑
[00051])。
[0024]
图4显示了筛查和诊断怀疑患有前列腺癌的患者的示意图。
[0025]
患者首先提供尿样用于三层sentinel
tm
分析。提取总尿液外来体rna并询问以下的表达:特定用于sentinel
tm
pca测试的sncrna(seq id no:1
‑
280);特定用于sentinel
tm
cs测试的sncrna(seq id no:281
‑
560);以及特定用于sentinel
tm
hg测试的sncrna(seq id no:561
‑
840)。表达特征将用于将患者分为前列腺癌患者和无前列腺癌患者(sentinel
tm
pca测试,第1层)。负分数的患者将每12个月返回进行监测。
[0026]
在第二层中,具有正sentinel
tm
pca分数的患者(患有前列腺癌的患者)将接受使用sentinel
tm
cs测试的二次分析,其将他们分类为临床无意义(gg1)肿瘤或临床有意义的肿瘤(gg2
‑
gg5)。将建议对临床无意义(gg1)肿瘤的患者进行主动监督(as),并通过每季度sentinel
tm
cs测试进行持续监测,以确定肿瘤没有进展到gg2或更高。具有临床意义的肿瘤(gg2
‑
gg5)的患者将被转诊进行立即治疗。
[0027]
对于某些患者,第三个分类层sentinel
tm
hg测试将进一步将患者分类为具有gg1
‑
gg2肿瘤或gg3
‑
gg5肿瘤。该测试旨在识别需要立即干预的gg3
‑
gg5癌症患者。gg1或gg2患者可以通过每季度sentinel
tm
hg测试进行监测,以识别进展为gg3并因此需要治疗干预的患
者。sentinel
tm
cs和sentinel
tm
hg的可用性为患者和医疗保健提供者都提供了用于做出治疗决策的个性化信息。
[0028]
图5显示了发现pca实验的输出。pca发现研究使用非常仔细定义的患者群组[nepc=89;癌症(gg1
‑
gg5)=146],其具有充分表征的组织病理学,以鉴定在mir4.0微阵列上询问的6,599个sncrna中信息量最大的序列。
[0029]
图5(左小图):训练数据集中无癌症(nepc)和癌症(gg1
‑
gg5)状态的散点图。正的发现pca分数表示患有前列腺癌,而负的发现pca分数表示没有癌症。由核心活组织检查的组织病理学确定的癌症状态以蓝色(无癌症)和红色(癌症)圆圈显示。
[0030]
图5(右小图):用于sentinel
tm
pca测试识别信息量最大sncrna实体的信息性sncrna的识别(显示前35个,每个圆圈代表一个实体),用于使用专有选择算法区分无癌症和癌症状态。产生的sentinel
tm
pca测试询问了280个sncrna,包括145个信息量最大的sncrna序列,其包括60个snorna和85个mirna实体,如条形图所示。绿色:mirna实体;黄色:snorna实体。
[0031]
图6(左小图)显示了发现cs实验的输出。cs发现研究使用非常仔细定义的患者群组[gg1=90;gg2
‑
gg5=56],其具有充分表征的组织病理学,以鉴定在mir4.0微阵列上询问的6,599个sncrna中信息量最大的序列。正的发现cs分数表示患有gg2
‑
gg5癌症(黄色圆圈),而负的发现cs分数表示患有gg1癌症(绿色圆圈)。
[0032]
图6(右小图):用于sentinel
tm
cs测试识别信息量最大sncrna实体的信息性sncrna的识别(显示前35个,每个圆圈代表一个实体),用于使用专有选择算法区分gg1和gg2
‑
gg5癌症状态。产生的sentinel
tm
cs测试询问了280个sncrna,包括145个信息量最大的sncrna序列,其包括66个snorna和130个mirna实体,如条形图所示。绿色:mirna实体;黄色:snorna实体。
[0033]
图7(左小图)显示了发现hg实验的输出。hg发现研究使用非常仔细定义的患者群组[gg1 gg2=181;gg3
‑
gg5=55],其具有充分表征的组织病理学,以鉴定在mir4.0微阵列上询问的6,599个sncrna中信息量最大的序列。正的发现hg分数表示患有gg3
‑
gg5癌症(紫色圆圈),而负的发现hg分数表示患有gg1 gg2癌症(棕色圆圈)。
[0034]
图7(右小图):用于sentinel
tm
hg测试的信息性sncrna的鉴定。使用专有选择算法识别信息最丰富的sncrna实体(显示前35个,每个圆圈代表一个实体),用于区分gg1和gg2
‑
gg5癌症状态。产生的sentinel
tm
cs测试询问了280个sncrna,包括196个信息量最大的sncrna序列,其包括66个snorna和130个mirna实体,如条形图所示。绿色:mirna实体;黄色:snorna实体。
[0035]
图8a
‑
8c显示了使用sentinel
tm
pca测试对尿液外来体sncrna进行高通量openarray
tm
询问的临床验证。显示了来自1436名男性(用于交叉验证在发现pca阶段鉴定的sncrna询问的训练组中的836名受试者和在验证研究中使用的600名独立受试者)的病例
‑
对照研究的数据。
[0036]
图8a:检查600名患者(300名无癌症;300名癌症)的验证组中癌症状态的散点图。无癌症(黑色圆圈)和癌症(绿色圆圈)患者的分类,其中正的sentinel
tm
pca分数表示患有前列腺癌,而负的sentinel
tm
pca分数表示没有癌症。
[0037]
图8b:检查600名患者的验证组中癌症状态的排序图。无癌症(黑色圆圈)和癌症
(绿色圆圈)患者的分类,其中正的sentinel
tm
pca分数表示患有前列腺癌,而负的sentinel
tm
pca分数表示没有癌症。
[0038]
图8c:sentinel
tm
pca测试的接收者操作曲线(roc)。图8a和8b中显示的测试组中600名患者的分析的roc曲线是通过对不同的用户定义的假阴性率连续计算(1
‑
特异性)来计算的。表6中报告的性能特征(参见[000112])来自用户定义的0.05假阴性率(以红色显示)。
[0039]
图9a
‑
9c显示了使用sentinel
tm
cs测试对尿液外来体sncrna进行高通量openarray
tm
询问的临床验证。显示了来自1436名男性(用于交叉验证在发现cs阶段鉴定的sncrna询问的训练组中的836名受试者和在验证研究中使用的600名独立受试者)的病例
‑
对照研究的数据。
[0040]
图9a:检查300名前列腺癌患者(146名gg1
‑
低等级和154名gg2
‑
gg5中和高等级)的验证组中癌症状态的散点图。低等级(青色圆圈)以及中和高等级癌症(橙色圆圈)患者的分类,其中正的sentinel
tm
cs分数表示患有高等级前列腺癌,而负的sentinel
tm
cs分数则表示为低等级癌症。
[0041]
图9b:检查300名前列腺癌患者的如图9a所示的验证组中癌症状态的排序图。低等级(青色圆圈)以及高等级癌症(橙色圆圈)患者的分类,其中正的sentinel
tm
cs分数表示患有高等级前列腺癌,而负的sentinel
tm
cs分数则表示为低等级癌症。
[0042]
图9c:sentinel
tm
cs测试的接收者操作曲线(roc)。如图9a和9b所示的对300名前列腺癌患者进行分析的roc曲线是通过对不同的用户定义的假阴性率连续计算(1
‑
特异性)来计算的。表6中报告的性能特征(参见[000112])来自用户定义的0.05假阴性率(以红色显示)。
[0043]
图10a
‑
10c显示了使用sentinel
tm
hg测试对尿液外来体sncrna进行高通量openarray
tm
询问的临床验证。显示了来自1436名男性(用于交叉验证在发现hg阶段鉴定的相同sncrna询问的训练组中的836名受试者和在验证研究中使用的600名独立受试者)的病例
‑
对照研究的数据。
[0044]
图10a:检查300名前列腺癌患者(200名gg1 gg2低等级和100名gg3
‑
gg5中和高等级)的验证组中癌症状态的散点图。低等级(青色圆圈)以及中和高等级癌症(橙色圆圈)患者的分类,其中正的sentinel
tm
cs分数表示患有高等级前列腺癌,而负的sentinel
tm
cs分数则表示为低等级癌症。
[0045]
图10b:检查300名前列腺癌患者的如图10a所示的验证组中癌症状态的排序图。低等级(蓝色圆圈)以及高等级癌症(红色圆圈)患者的分类,其中正的sentinel
tm
hg分数表示患有高等级前列腺癌,而负的sentinel
tm
hg分数则表示为低等级癌症。
[0046]
图10c:sentinel
tm
hg测试的接收者操作曲线(roc)。如图10a和10b所示的对300名前列腺癌患者进行分析的roc曲线是通过对不同的用户定义的假阴性率连续计算(1
‑
特异性)来计算的。表6中报告的性能特征(参见[000112])来自用户定义的0.05假阴性率(以红色显示)。
具体实施方式
[0047]
可以通过参考形成本公开的一部分的以下详细描述更容易地理解本主题。应当理
解,本发明不限于本文描述和/或示出的具体产品、方法、条件或参数,并且所用的术语仅用于以举例的方式描述具体的方面和实施方式的目的,并且并不旨在限制所要求保护的发明。
[0048]
本发明涉及一种在受试者中筛查、诊断和治疗前列腺癌的方法。该方法提供了稳健的测试,以便(1)对未知前列腺癌状态的男性患者进行分类和(2)准确区分来自患者的生物样品中的前列腺癌等级。该方法基于对来自患者生物样品的sncrna集合的聚集表达谱的检测和关联,以使用sentinel
tm
pca测试确定患者是否患有前列腺癌。对于鉴定为患有前列腺癌的患者,进一步询问外来体sncrna,其使用sentinel
tm
临床有意义(cs)测试,以区分具有临床意义或侵袭性(gg2
‑
gg5)的患者与临床上无意义或惰性(gg1)前列腺癌的患者,以及sentinel
tm
高等级(hg)测试,以识别高等级、高风险(gg3
‑
gg5)前列腺癌患者。
[0049]
所公开的方法基于一种无偏统计方法,该方法被开发以鉴定与感兴趣的表型最相关的单个序列和序列组合的重要相互作用。该方法基于(i)mirna对mrna的调节作用和(ii)snorna通过核糖体rna、trna和其他核rna的转录后修饰对mrna可译性的影响,这产生改变蛋白质功能和表型的新蛋白质产物。
[0050]
所公开的计算/统计方法分析尿外来体sncrna以提供对sncrna之间的关键关联的非常精细的分析,这导致鉴定准确预测前列腺癌表型的sentinel序列。附图3对此进行了说明。例如,在单一实体分析中,单个sncrna的表达水平与前列腺癌的等级组(表型)相关。对于每个sncrna实体,有两个信息结果:实体的表达水平相对于对照病理(例如,无癌症)的增加或表达水平的降低。两种表型之间的表达没有变化表明(1)与任一表型没有关联,并且(2)该实体不能用作任一表型的标志物。因此,当在分析中使用单个实体时,只有两种信息结果,使得所有可能的sncrna相互作用都没有被探索。
[0051]
在两个实体分析中,检查两个实体之间所有可能相互作用的表达变化的关联产生8个不同的信息结果和1个非信息结果(当两个实体的表型均未差异表达时)(参见图3“2个实体的询问”)。因此,在sentinel
tm
测试的背景下,当比较两个sncrna实体的所有可能组合与特定等级组的关联时,有8种不同的方式将导致sncrna对和等级组之间有意义的关联。这提供了更详细的分析,其揭示了sncrna表达水平与等级分组之间的隐藏关联。
[0052]
使用三个或四个或更多sncrna实体的相同方法可以对sncrna表达和表型(等级分组)之间的关联进行非常精细的分析,从而可以评估未知疾病状态的患者并使用算法选择的尿外来体sncrna表达水平预测个体疾病状态。
[0053]
sentinel
tm
pca和sentinel
tm
hg测试sentinel
tm
cs平台的开发
[0054]
sentinel
tm
pca测试是一种分类平台或算法,其基于对特征sncrna(即mirna和snorna序列)水平集合的分析。每个序列的预测值是通过数据驱动的选择算法定义的,该算法独立于序列在前列腺生物学中的先验确定的生物学作用。选择算法在数据集上进行训练,该数据集由以下组成:(1)在泌尿科就诊的与前列腺癌无关的状况的对照受试者;(2)基于活检结果已知没有前列腺癌的疑似前列腺癌受试者;和(3)诊断为前列腺癌且其核心针活检组织病理学报告为等级组1至5(gg1
‑
gg5)的患者。
[0055]
为了建立用于sentinel
tm
测试的强大数据集,使用affymetrix mir 4.0微阵列询问从这些患者训练集的尿液外来体中获得的外来体sncrna,以限定表达特征。这些使用具有充分表征的组织病理学的选定受试者的研究被称为发现pca、发现cs和发现hg测试。纳入
“
无癌症”组的患者是从因与泌尿科肿瘤学无关的问题而在泌尿科诊所就诊的年龄匹配的男性以及接受过一次或多次12针诊断核心针活检而显示无前列腺癌证据(nepc)的男性中精心挑选的。对于“癌症”群组中的患者,对每个肿瘤的核心针活检的病理等级组分类进行了全面评估。这些精心挑选的患者组(无癌症和处于不同癌症阶段的癌症组)构成了发现pca、cs和hg测试开发中的训练集。用于发现实验的235名患者的人口统计数据如表4所示(参见[000103]
‑
[000104])
[0056]
选择算法
[0057]
使用选择算法识别出区分癌症和无癌症的信息量最大的sncrna序列,该算法确定哪些sncrna序列在未患有前列腺癌(nepc)的患者和患有前列腺癌(gg1
‑
gg5)的患者之间存在差异。下面举例说明了发现pca测试。选择算法在具有精心定义的病理的大群参与者[89名nepc受试者和146名癌症患者(gg1
‑
gg5)]中测试尿液外来体sncrna的水平与疾病病理分期(癌症/非癌症)的相关程度。选择算法单独评估在mir4.0阵列上询问的6,599个sncrna中的每一个与已知肿瘤病理的相关程度。由于许多sncrna被协调调节,因此该算法然后评估2个sncrna、3个sncrna或4个sncrna的所有组合,然后使用留一法策略检查每个单独sncrna,以评估每个单独sncrna在该疾病的病理学中的重要性。正如预期的,从选择算法中排除6,599个sncrna序列中的大部分对区分患有前列腺癌和没有前列腺癌没有影响,因为它们与任一种病理都没有差异关联。sncrnas评估的影响可以使用图5(右小图)中显示的重要性图进行可视化。该重要性图显示以下内容:(1)一些外来体sncrna在不同病理中以不同水平存在,(2)sncrna是snorna和mirna,表明一种类型的sncrna不足以进行所公开的分析,以及(3)使用该算法诊断在分类评估中不会有超过280个sncrna序列的改变。
[0058]
发现cs测试(其区分低风险(gg1)和中高风险前列腺癌(gg2
‑
gg5))的信息序列是使用适当的等级组和相同的选择策略确定的(图6)。用于该分析的患者群体包括89名nepc受试者和146名癌症患者(gg1
‑
gg5)]
[0059]
发现hg测试(其区分低和中风险(gg1 gg2)和高等级、高风险(gg3
‑
gg5)前列腺癌)的信息序列也被类似地确定(图7)。用于该分析的患者群体包括181名gg1 gg2癌症患者和55名gg3
‑
gg5癌症患者。(图7)。
[0060]
重要的是要注意,虽然一些sncrna在测试之间是常见的,但它们在疾病状态分类中的相对重要性因测试(即,发现pca测试、cs测试和hg测试)而异。
[0061]
对于每个测试,信息最丰富的280个sncrna seq.id no:1
‑
840)用于设计定制的openarray
tm
平台。用于每个sentinel
tm
测试的openarray
tm
平台在1436名患者的大型病例
‑
对照研究中得到了进一步验证。用于训练和验证sentinel
tm pca、sentinel
tm
cs和sentinel
tm
hg测试的受试者的人口统计数据示于表5(见[000109]
‑
[000110])。选择了600名受试者的分层随机样品来识别验证数据集;其余836名受试者作为训练数据集。对600名患者的验证样品进行分层,以便相同数量的受试者活检阴性与活检阳性(各300名),并且活检阳性病例中200名为gg1 gg2(146名gg1和54名gg2)和100名gg3
‑
gg5。
[0062]
用于识别前列腺癌的sentinel
tm
pca测试
[0063]
sentinel
tm
pca、sentinel
tm
cs和sentinel
tm
hg测试基于分类算法,该算法将每位患有未知疾病状态的患者的sncrna表达特征作为输入并产生sentinel
tm
分数;通过将此分数与预先确定的临界值(从训练数据集中的交叉验证获得)进行比较来对参与者进行分类,该
临界值将对未来疾病状态未知(但表达特征已知)患者进行分类的灵敏度控制在用户定义的水平(通常为95%或更高)。
[0064]
将sentinel pca分数与计算的临界值进行比较,该临界值将未来患者的灵敏度控制在期望水平,例如95%,以区分pca测试的有前列腺癌和无前列腺癌(图7)。sentinel
tm
pca测试使用280个sncrna(由发现pca测试识别),其中145个独特的sncrna:60个mirna和85个snorna是高度有信息的。这定义了用于将患者分为或不分为前列腺癌的分类边界。临界值由算法确定,使得将患者分为癌症/无癌症的sentinel
tm pca分数在20次中有19次正确地将患者分类为患有癌症(即,具有95%的灵敏度)。
[0065]
表1:用于pca测试分析的seq id no:1
‑
280
[0066]
[0067]
[0068]
[0069]
[0070]
[0071]
[0072]
[0073]
[0074]
[0075]
[0076]
[0077]
[0078]
[0079]
[0080]
[0081]
[0082]
[0083]
[0084]
[0085][0086]
sentinel
tm
cs检测用于识别低等级(惰性)前列腺癌
[0087]
sentinel
tm
临床意义(cs)测试使用类似的分类算法产生sentinel
tm
cs分数,并将其与计算的截止值进行比较。临界值将未来患者的灵敏度控制在期望水平(95%),以区分临床有意义的癌症(gg2
‑
gg5)(如果sentinel
tm
cs分数大于或等于临界值)和临床无意义癌症(gg1))(如果sentinel
tm
cs分数小于临界值)。该算法仅使用用于训练sentinel
tm
pca测试的数据集中已知患有前列腺癌的患者子集进行训练。同样,使用分类算法,使用280个sncrna作为基础来定义sentinel
tm
cs测试的表达特征。sentinel
tm
cs测试使用280个sncrna(由发现cs测试识别,其中135个独特的sncrna:130个mirna和66个snorna是高度有信息的。
[0088]
表2:在cs测试分析中使用的seq id no:281
‑
560
[0089]
[0090]
[0091]
[0092]
[0093]
[0094]
[0095]
[0096]
[0097]
[0098]
[0099]
[0100]
[0101]
[0102]
[0103]
[0104]
[0105]
[0106]
[0107]
[0108]
[0109][0110]
sentinel
tm
hg检测以识别高等级前列腺癌患者
[0111]
对于归类为有前列腺癌的个体,使用类似的方法来训练和验证sentinel
tm
高等级(hg),其区分gg1 gg2(低和有利的中风险癌症)和不利的中风险和高风险前列腺癌(gg3
‑
gg5)。这些确定的信息序列构成了sentinel
tm
hg测试的基础。其他分析表明,同一sncrna群组可用于将癌症患者从gg3
‑
5(高风险癌症)分至gg1 gg2(低和中风险癌症)中。这种生物统计分析构成了sentinel
tm
hg测试的基础,该测试利用了发现hg测试确定的280个sncrna,其中280个独特的sncrna:191个mirna和89个snorna是高度有信息的。
[0112]
表3:用于hg测试分析的seq id no:561
‑
840
[0113]
[0114]
[0115]
[0116]
[0117]
[0118]
[0119]
[0120]
[0121]
[0122]
[0123]
[0124]
[0125]
[0126]
[0127]
[0128]
[0129]
[0130]
[0131]
[0132]
[0133][0134]
sentinel
tm
pca、cs和hg测试中sncrna的选择独立于psa、gleason分数或生物通路分析,因此完全没有偏见。因为该算法是使用从独立训练集获得的sncrna水平进行验证的,该训练集由一组核心针活检呈阳性或阴性(pca测试)的参与者或标记为患有晚期疾病(gg3
‑
5)或对于cs测试未患有疾病(没有pca证据或gg1
‑
1)的患者组成。(见表4,[000103]
‑
[000104]和表5[000109]
‑
[000110]),这种统计方法最大限度地减少了第1类错误(假阴性)和第2类错误(假阳性),这确保了测试严格区分无癌症与低等级癌症、低与中等级癌症以及中与高等级疾病。基于分析中使用的算法,所描述的发明没有假阴性和非常低(<5%)的假阳性率。
[0135]
基于上述三项测试,openarray
tm
平台依次询问从尿液外来体中提取的单个sncrna样品中存在的信息性rna实体,而不会影响三项测试的灵敏度和特异性。
[0136]
在一个方面,本公开内容提供了一种用于诊断前列腺癌的方法,该方法包括允许人们区分临床有意义肿瘤和惰性肿瘤的平台,并且将基于被询问的子集sncrna的数据放入已基于独立训练数据集验证的算法中。
[0137]
一方面,用于诊断男性患者的前列腺癌的方法包括(1)从患者获得生物样品,(2)检测结合选自seq id no:1
‑
280的多个核酸或杂交探针的特征小非编码rna(sncrna)的集合的聚集表达谱;和(3)使用pca测试关联特征sncrna的集合的聚合表达谱以确定患者是否处于前列腺癌的风险中,即,没有前列腺癌证据或患有前列腺癌。
[0138]
在另一方面,本公开提供了一种使用该方法筛查前列腺癌的方法。在又一方面,本公开提供了一种用于预测受试者中前列腺概率的方法。
[0139]
对于确定有患前列腺癌风险的患者(即,确定患有前列腺癌),使用sentinel
tm
临床有意义(cs)测试重新分析样品以区分患有临床有意义或侵袭性前列腺癌(gg2
‑
gg5)的患者与临床无意义或惰性(gg1)前列腺癌患者。在一个实施方案中,当特征sncrna的多个或集合的聚集或组合表达谱高于或等于前列腺癌生物样品中的聚集表达谱时,患者被鉴定为患有侵袭性前列腺癌,或当特征sncrna的集合的聚集表达谱小于或等于低等级前列腺癌生物样品中的聚集表达谱时,鉴定受试者为患有低等级前列腺癌。
[0140]
在一个实施方案中,生物样品包括但不限于前列腺组织、血液、血浆、血清、尿液、尿液上清液、尿细胞沉淀、脑脊液、精液、前列腺分泌物和前列腺细胞。在一些实施方案中,生物样品是尿样。在又一个实施方案中,样品是从尿样中分离的外来体。在一个优选的实施方案中,样品是从来自尿样的外来体分离的sncrna。
[0141]
外来体是起源于真核细胞内体区室的小细胞外囊泡(ev)。它们存在于生物体液中,包括血液、尿液、精液和脑脊液。外来体的生物起源还不是很清楚;然而,普遍认为它们出现在早期内体途径分叉以形成晚期内体和多囊泡内体的点上,这是外来体途径的第一阶段。外来体包含sncrna,其包括mirna和小核仁rna(snorna),它们分别来自细胞的细胞质和核仁区域。肿瘤微环境中外来体和ev的存在与许多肿瘤类型的恶性肿瘤有关,包括前列腺癌和其他癌症。
[0142]
在某些实施方案中,从外来体分离的sncrna源自精液、血液、前列腺分泌物和脑脊液。在进一步的实施方案中,外来体从包括前列腺癌细胞的癌细胞、淋巴细胞和来自前列腺组织的细胞中分离。
[0143]
分离外来体的方法是本领域众所周知的,可以使用试剂盒例如外来体rna分离试剂盒(norgen biotek corp.,ontario,ca)进行。sncrna产量可以通过荧光法(qubit,thermo fisher scientific)量化,并且使用agilent 2100生物分析仪评估分离的sncrna的质量。
[0144]
从分离的外来体中提取的rna是小rna的混合物,统称为小非编码rna(sncrna),其包括mirna、snorna、scarna、sirna、snrna和exrna。由于它们的大小(<200个核苷酸),sncrna容易从生物样品中提取,包括,例如,福尔马林固定石蜡包埋(ffpe)组织或尿液。sncrna在固定或提取过程中不会降解,避免了从ffpe组织中提取mrna的固有问题。来自生物样品的约10ng sncrna的产量足以使用上文公开的norgen的外来体rna分离试剂盒进行多次分析。
[0145]
提取的sncrna被逆转录成cdna,它比rna更稳定,从而允许更长时间的储存。得到的cdna与选定的一组或一集合的特征sncrna探针或基因组阵列或微阵列芯片杂交,例如mir 4.0阵列(thermofisher scientific),用于进一步分析。sncrna探针信息集的选择与psa、gleason分数或生物途径无关。选定的一组或集合的特征sncrna包含seq id no:1
‑
280、281
‑
560和561
‑
840。集合中sncrna序列或探针的数量为145
‑
196个,优选不超过280个sncrna序列,以便放入sentinel
tm
pca、sentinel
tm
cs和sentinel
tm
hg测试中。通过将多达280个sncrna序列添加到算法中,测试更加精确。在分析中添加超过280个sncrna序列不会提高检测的精确度。
[0146]
实时pcr或rt
‑
pcr,通常称为qpcr或rt
‑
qpcr,用于量化目标序列的绝对量或比较样品之间目标序列的相对量。rt
‑
qpcr通过扩增过程中发出的目标特异性(探针)荧光信号
实时监测目标的扩增。尽管使用针对目标的序列特异性探针,但大多数rt
‑
qpcr反应期间都发生背景荧光,因此可以通过考虑实时pcr中的两个值来解决背景荧光信号的问题:(1)阈值线(c
t
)和(2)循环量化(c
q
)值。阈值线(c
t
)是当反应达到高于背景水平的荧光强度时的检测水平,即反应曲线开始指数期的点(拐点)。c
q
或循环定量值是样品反应曲线与阈值线相交处的pcr循环数。因此,c
q
值表示从样品中检测到真实信号需要多少个循环,即事件发生的时间,其中事件是荧光的饱和度,表示检测的最大水平。因为rt
‑
qpcr运行为每个样品提供了反应曲线,所以会有很多c
q
值。pcr循环仪中的软件将计算每个样品的c
q
值并绘制其图表。数值与样品中目标核酸的量成反比,并与样品中的目标拷贝数相关。较低的c
q
值(通常低于29个循环)表示大量的目标序列。相反,较高的c
q
值(超过38个循环)表明样品中目标核酸的量较低。然而,当反应曲线的斜率变为零时可以获得事件发生时间值,而不是在斜率最大时使用c
q
。在一个实施方案中,使用rt
‑
qpcr询问sncrna。在另一个实施方案中,使用qpcr询问sncrna。在进一步的实施方案中,按照制造商的说明使用affymetrix genechip
tm mirna 4.0阵列询问从尿外来体分离的sncrna。
[0147]
这些从训练集中获得的信息序列(通常为280个序列)然后被传输到openarray平台。然后在openarray平台上询问状态未知的患者样品,并使用分类算法确定sentinel分数。患者的状态由sentinel分数确定。
[0148]
在一个实施方案中,来自未知前列腺癌患者的sncrna水平在openarray平台上被询问,然后来自未知疾病状态的患者的sentinel分数可以与训练集的分数比较以确定患者的状态。
[0149]
在一个实施方案中,使用例如通过affymetrix genechip
tm
mirna 4.0阵列上的rt
‑
qpcr分析来自未知前列腺疾病状态的患者的测试样品(测试样品)中的sncrna水平获得的数据可以与来自健康患者(没有癌症证据)或从患有前列腺癌的受试者身上获得的健康细胞的数据进行比较。从测试样品的sncrna水平分析中获得的数据也可以与通过分析健康(无癌症)和非健康(患有泌尿生殖系统癌症)患者建立的临床基线进行比较,并且非健康患者进一步分类为不同的特定癌症类型,其可进一步分类为特定癌症类型(例如前列腺癌等)的不同阶段或严重程度以及特定疾病的不同阶段。在一个实施方案中,将在affymetrix genechip
tm
mirna 4.0阵列上使用rt
‑
qpcr分析测试样品中的sncrna表达所获得的数据与来自健康患者(无癌症证据)的数据进行比较,或者也可以将数据与通过分析健康(无癌症)和非健康(患有前列腺癌)患者建立的临床基线进行比较,并且将非健康患者进一步分类为前列腺癌的不同阶段(gg1和gg2
‑
gg5或gg1 2和gg3
‑
gg5)。
[0150]
在一些实施方案中,该方法使用openarray
tm
技术(thermofisher scientific)来询问一组sncrna(例如,mirna、snorna)。openarray
tm
技术使用具有48个子阵列的显微镜载玻片大小的板。每个子阵列有64个通孔,每个孔的直径为300μm,深度为300μm。将孔用亲水和疏水涂层处理,以便通过表面张力将试剂保留在通孔中。openarray
tm
技术具有3,072个通孔(48x64),提供了使采用大量样品、分析或两者的实时pcr研究效率更高的系统。因此,该系统允许使用微量的样品和试剂在短时间内处理大量用于基因表达的样品。该方法采用一种算法,该算法依赖于每个sncrna的表达水平和活检(至少12个核心针活检)的分级。在前列腺癌的情况下,该方法独立于血清前列腺特异性抗原(psa)水平、gleason分数(两者都不是肿瘤进展的有意义的标志物)或患者年龄。该方法还独立于对生物途径的任何分析。本方
法使用从受试者(例如,尿样、尿外来体或前列腺组织样品)分离的sncrna将男性分为患有前列腺癌(惰性(无临床意义)或侵袭性(有临床意义))和没有前列腺癌的人群。这种方法可以取代血清psa作为前列腺癌的主要筛查方法。
[0151]
一方面,本公开提供了一种方法,该方法基于经过独立于病理学(gleason分数)、肿瘤体积或psa的分类算法的所询问的特征sncrna集合的聚集表达谱来区分具有临床意义的前列腺癌。从具有已知癌症结果的患者的生物样品中提取的rna被逆转录并与包含sncrna的全基因组阵列(例如,affymetrix genechip mir 4.0)杂交。鉴定了在具有临床意义的前列腺肿瘤中差异调节的小非编码rna。在一个实施方案中,将鉴定与seq id no:281
‑
561的探针杂交的sncrna的来自开放阵列的信号的绝对值与在具有临床意义(gg2
‑
5)前列腺癌肿瘤中发现的聚集表达谱进行比较。在另一个实施方案中,将鉴定与seq id no:561
‑
840的探针杂交的sncrna的来自开放阵列的信号的聚集值与在临床上低和有利的中等级(gg1 gg2)中发现的绝对表达谱进行比较,这相对于不利的中和高等级(gg3
‑
gg5)前列腺癌肿瘤。
[0152]
所公开的方法在从收到尿样到获得sentinel分数的72
‑
96小时内提供了对前列腺癌预后的稳健且准确的确定。在另一个实施方案中,将鉴定的与seq id no:281
‑
560和561
‑
840结合的sncrna的聚集表达谱与具有临床意义的前列腺癌中sncrna的聚集表达谱进行比较。在另一个实施方案中,将鉴定的与seq id no:281
‑
560和561
‑
840结合的经询问的sncrna的聚集表达谱与具有临床意义的前列腺癌中sncrna的聚集表达谱进行比较。在进一步的实施方案中,将鉴定的与seq id no:281
‑
560和561
‑
840结合的sncrna的聚集表达谱与具有临床意义的前列腺癌样品中sncrna的聚集表达谱进行比较。因此,可以启动适当的治疗选择(或缺乏治疗选择)。
[0153]
术语相对聚集表达谱可互换使用。将至少多个sncrna的聚集表达谱组合并与具有临床意义的前列腺癌组织中的相同聚集表达谱进行比较。在一些实施方案中,至少40个sncrna被组合并与具有临床意义的前列腺癌组织中的相同聚集表达谱进行比较。在一些实施方案中,至少90个sncrna被组合并与具有临床意义的前列腺癌组织中的相同聚集表达谱进行比较。在一些实施方案中,至少150个sncrna被组合并与具有临床意义的前列腺癌组织中的相同聚集表达谱进行比较。在一些实施方案中,至少200个sncrna被组合并与具有临床意义的前列腺癌组织中的相同聚集表达谱进行比较。在一个优选的实施方案中,将至少224个sncrna和不超过280个sncrna组合并与具有临床意义的前列腺癌组织中的相同聚集表达谱进行比较。在某些实施方案中,与低等级前列腺癌组织中的聚集表达谱相比更高的聚集表达谱表明患者患有侵袭性前列腺癌并且需要治疗。在其他实施方案中,等于或低于低等级前列腺癌组织中的聚集表达谱的聚集表达谱表明患者没有侵袭性前列腺癌并且可能需要监测但不需要治疗。
[0154]
在一些实施方案中,所选sncrna的聚集表达谱是sncrna的各种类型的调节表达的聚集。相对于其他组织/肿瘤类型(例如健康前列腺组织、低等级前列腺癌组织或高等级前列腺癌组织)中的相同sncrna,调节的表达可以是降低或增加的表达谱。
[0155]
在其他实施方案中,所选sncrna的聚集表达谱可以是同一组织样品中某些sncrna的降低的聚集表达谱的聚集以及其他sncrna的增加的聚集表达谱的聚集。例如,特征sncrna集合的进展分数或聚合表达谱可以包括具有相对于另一组织类型或同一组织样品
中的其他sncrna降低的聚合表达谱的一个或多个sncrna,而一个或多个剩余的sncrna相对于同一组织样品中的另一组织类型或其他sncrna表现出增加的聚合表达水平。不同调节的sncrna集合的聚集表达谱提供了一个复杂的、无偏见的前列腺肿瘤是否具有临床意义的指示。与仅评估与正常组织相比单个靶分子的存在或不存在或简单增加或减少的其他方法不同,所公开的方法提供了对前列腺组织样品的真正无偏的、独立和多变量的分析,从而允许令人惊讶地准确诊断前列腺癌肿瘤是否具有临床意义。
[0156]
在一些方面,该方法提供了特征sncrna集合的聚集表达谱用于监测转移和癌症分期的用途。
[0157]
在另一方面,本公开提供了一种用于基于使用公开的分类算法询问和进行分析的特征sncrna集合的聚集表达谱来检测泌尿系统恶性肿瘤的方法。在一些实施方案中,恶性肿瘤是前列腺的癌症。
[0158]
本公开提供了一种基于算法的分子诊断分析,用于预测前列腺癌患者的临床结果。一种或多种sncrna的表达水平可以单独使用或排列成功能基因子集,以计算可用于预测临床结果可能性的定量分数。
[0159]“定量分数”是算术或数学计算的数值,用于帮助简化或公开或告知更复杂的定量信息的分析,例如公开的sncrna或sncrna子集的某些表达谱与前列腺癌患者的临床结果的可能性的相关性。可以通过应用特定算法来确定定量分数。在所公开的方法中用于计算定量分数的算法可以对sncrna的表达谱值进行分组。sncrna的分组可以至少部分地基于根据生理功能或组分细胞特征的sncrna的相对贡献的知识进行,例如在本文讨论的组中。可以确定sncrna组的定量分数(“sncrna组分数”或sentinel
tm
分数)。此外,组的形成可以促进基因或基因子集的各种聚集表达谱对定量分数的贡献的数学加权。代表生理过程或组成细胞特征的sncrna或sncrna组的权重可以反映该过程或特征对癌症病理学和临床结果例如癌症的复发或升级/升期的贡献。本发明提供了多种用于计算定量分数的算法。例如,本公开中的分类算法以相同的方式工作以开发用于区分不同疾病状态的sentinel分数。分类算法从训练数据集中为临床有意义和无意义的疾病状态选择不同的sncrna序列。
[0160]
另一方面,选择算法测试了尿液外来体sncrna的聚集表达谱与疾病的病理阶段(癌症/无癌症)在具有已知病理的大群参与者中的相关性。选择算法单独评估在mir4.0阵列上询问的6,599个sncrna中的每一个与参与者已知病理的相关程度。然后,它反复评估mir 4.0阵列询问的6,599个sncrna中2个sncrna、3个sncrna或4个sncrna的所有组合,然后使用留一法策略检查每个单独sncrna,以评估每个单独sncrna在该疾病的病理学中的重要性。然后通过使用开放阵列(open array)询问选定的sncrna确定具有未知疾病状态的患者的sentinel分数,并通过将分数与训练数据集的分数进行比较来确定临床状态。在本发明的一个实施方案中,定量分数的增加表明阴性临床结果的可能性增加。
[0161]
基于定量分数和累积或绝对或聚集表达谱,还可以决定治疗方法。治疗前列腺癌的方法包括用于完全手术切除前列腺组织的手术、施用有效剂量的放射和施用治疗有效量的用于治疗前列腺癌的药物,或以上的组合。
[0162]
通过实施本发明的方法提供的基于算法的测定和相关信息促进了前列腺癌的最佳治疗决策。例如,这样的临床工具将使医生能够识别患有侵袭性癌症的可能性低的患者,因此除了每3个月、6个月或12个月的常规随访或主动监测外,不需要进一步的医疗干预。没
有癌症的患者不需要每年一次的医疗干预返回进行随访。有发展为侵袭性癌症风险的患者需要医学干预,包括但不限于用以下治疗:一种或多种化学治疗剂(例如泰素帝、卡巴他赛、多西他赛、米托蒽醌、表柔比星、紫杉醇和雌莫司汀等)、激素疗法(例如促黄体激素释放激素激动剂以防止睾酮的产生,例如亮丙瑞林、戈舍瑞林和曲普瑞林,或防止睾酮到达癌细胞的抗雄激素药物,例如比卡鲁他胺和尼鲁他胺)、免疫疗法、放射、冷冻疗法、手术或其组合。
[0163]
使用所公开的方法监测接受治疗的患者以确定患者对治疗的反应。一方面,本公开提供了一种用于确定患者对治疗的反应的方法,包括:(i)从患者获得生物样品,(ii)检测来自生物样品的小非编码rna(sncrna)的特征集合的聚集表达谱,其中sncrna的集合包含seq id no:1
‑
280、281
‑
560和561
‑
840,(iii)通过将seq id no:1
‑
280、281
‑
560和561
‑
840的聚集表达谱与治疗前的进行比较,关联治疗后受试者的seq id no:1
‑
280、281
‑
560和561
‑
840的sncrna的聚集表达谱,(iv)确定患者是否对治疗有反应以及是否有修改治疗的需要。在一个实施方案中,该方法进一步比较来自上述(iii)的sncrna的特征集合的所得聚集表达谱,然后与来自患有具有已知等级组的前列腺的目标人群的大训练数据集的sncrna的特征集合的聚集表达谱进行比较,以确定(a)患者前列腺癌是否稳定(与等级组相比没有明显变化),(b)患者对治疗是否有反应,即患者好转(结果显示肿瘤类似于具有较低等级组的肿瘤),或(c)患者无反应(当结果显示肿瘤类似于较高等级组的肿瘤时,患者变得更糟),以上基于特征sncrna集合的聚集表达谱和该等级组的sentinel分数,以及是否有修改治疗的需要。治疗修改包括但不限于调整化学治疗剂、放射、免疫治疗剂或激素的施用浓度或用量,添加或去除所用的一种或多种药剂。
[0164]
在另一方面,本公开提供了一种方法,其通过比较seq id no:1
‑
280、281
‑
560和561
‑
840在训练数据集和患者的早期资料中的聚集表达谱,基于包含seq id no:1
‑
280、281
‑
560和561
‑
840的特征sncrna集合的聚集表达谱来确定疾病复发、疾病进展或存活可能性。
[0165]
在另一方面,本公开提供了一种系统,其用于确定患者是否患有癌症或患有癌症并将患有癌症的受试者分类为(i)惰性(低等级,gg1)、(ii)中或高等级(gg2
‑
gg5)、(iii)低/中风险(gg1
‑
gg2)或(iv)侵袭性(高等级,gg3
‑
gg5)前列腺癌,所述系统包括至少三个处理器,其配置为(a)询问sncrna序列的信息序列,(b)确定和比较sentinel分数以确定受试者是否患有前列腺癌或没有前列腺癌,并将确定患有癌症的受试者分类到各个等级组,例如低等级、中/高等级、低/中风险或侵袭性等级癌症。确定没有癌症证据的受试者不需要医疗干预,并将每年返回进行一次随访。确定患有低等级或低/中等级前列腺癌的受试者除了每3、6或12个月的常规随访或主动监测外不需要医疗干预,以及确定患有中/高等级或侵袭性前列腺癌的受试者需要医疗干预。
[0166]
实施例
[0167]
本发明的这一方面和其他方面通过以下非限制性实施例进一步说明。
[0168]
实施例1
[0169]
研究群体:
[0170]
两个独立的患者群组用于开发和验证sentinel
tm
pca和sentinel
tm
cs测试。用于开发sentinel
tm
pca以将患者分类为患有癌症或没有癌症的233名参与者的临床和人口统计学特征是基于对特征snrna集合的统计分析。对于分类为患有癌症的患者,使用sentinel
tm
cs
测试将gg1(惰性、低风险癌症)患者与gg2
‑
5(分别为中、高风险和侵袭性癌症)区分开来,这也是基于使用第二分类算法统计分析特征sncrna的另一集合以将肿瘤分类为gg1与gg2
‑
5。两个测试中的sncrna都由affymetrix mir 4.0阵列询问。
[0171]
尿液收集和处理
[0172]
用于开发sentinel
tm
pca和cs测试以及基于us的回顾性研究群组的尿液样品在访问当天在两个临床地点收集进行临床检查:albany medical center(albany,ny,usa)和suny downstate medical center(brooklyn,ny,usa)。用于回顾性研究的剩余样品从加拿大多伦多gubiobank,university health network索取,并在
‑
20℃时散装冷冻运输到mir科学实验室。经机构审查委员会批准,在每个参与点收集并匿名化患者信息。前列腺癌的诊断是通过核心针活检的组织病理学分级获得的;每个核心的肿瘤百分比和阳性核心的数量用于评估等级组(gg)。
[0173]
离心尿液样品以去除游离细胞和碎片。根据制造商的说明,使用外来体rna分离试剂盒(norgen biotek,on)提取rna。sncrna产量通过荧光测定法(qubit,thermo fisher scientific)定量,并将rna样品储存在
‑
80℃直到分析。
[0174]
总外来体sncrna的微阵列分析
[0175]
按照制造商的说明,使用affymetrix genechip
tm mir 4.0阵列询问sncrna。在这些阵列上分析的235名患者的符合maime的原始数据文件已存放在ncbi的gene expression omnibus中。(edgar r et al.gene expression omnibus:ncbi gene expression and hybridization array data repository.nucleic acid res 2002 30:207)。训练集中的6599个sncrna在affymetrix genechip
tm mirna 4.0阵列上询问。
[0176]
为每个参与者询问的小非编码rna实体使用专有的选择和分类算法进行分析。鉴定了用于区分癌症和非癌症受试者(seq id no:1
‑
280)以及等级组1和等级组2
‑
5患者之间的信息最丰富的序列。(seq id no:281
‑
842)
[0177]
基于quantstudio openarray
tm
的外来体sncrna询问
[0178]
cdna合成,选定mirna的预扩增:为了分析外来体mirna,按照制造商的建议,使用taqman
tm microrna逆转录试剂盒(thermo fisher scientific),在与三个特定mirna茎环引物库的单独反应中对总sncrna进行逆转录。mirna cdna库分别用pre
‑
amp引物库富集16个循环(95℃10分钟,55℃2分钟,72℃2分钟,95℃15秒,和60℃4分钟重复16个循环,99.9℃10分钟),并按照制造商的建议在quantstudio openarray
tm
上的三个56实体子阵列上进行询问。
[0179]
cdna合成、选定snorna的预扩增和询问:按照制造商的建议,使用高容量cdna逆转录试剂盒以单个pre
‑
amp引物库(thermo fisher scientific)对总sncrna进行逆转录。通过预扩增(95℃10分钟,95℃15秒和60℃4分钟,分别重复14和18个循环,以及99℃10分钟)富集snorna cdna产物,并在两个56实体子阵列中询问。
[0180]
统计分析:
[0181]
sentinel
tm pca测试基于分类算法,该算法已针对一组核心针活检呈阳性或阴性的参与者进行了训练。分类算法将患有未知疾病状态的参与者的sncrna表达特征作为输入,并产生sentinel
tm
分数;通过将sentinel
tm pca分数与预先确定的临界值进行比较对参与者进行分类,该临界值将对未来疾病状态未知(但表达特征已知)患者进行分类的灵敏度
保持在用户定义的水平(95%或更高)。第二种分类算法sentinel
tm cs测试操作类似于sentinel
tm pca测试。然而,sentinel
tm cs测试的分类算法在标记为低等级(gg1)的患者群组和标记为有利中至高等级前列腺癌(gg2
‑
gg5)的患者群组上训练。第三个分类算法sentinel
tm hg测试在确定为低风险和有利中风险(gg1 gg2)前列腺癌的患者群组和表征为不利中风险和高风险(gg3
‑
gg5)的第二群组上训练。
[0182]
sentinel
tm
测试范例在两层或三层中运行。首先,它使用参与者尿液中的sncrna特征以输入到sentinel
tm pca测试的分类规则中,以确定是否存在癌症;其次,对于确诊为癌症的那些患者,sentinel
tm cs测试确定癌症是否为低风险(gg1);第三,sentinel
tm
hg测试确定肿瘤是否为不利中或高风险(gg3
‑
gg5)。(见图4)
[0183]
表4:用于开发分类算法的群组的人口统计学和临床特征。
[0184][0185]
*
在豁免研究状态下,没有癌症证据的患者无法获得psa水平。
[0186]
表4建立了用于开发sentinel
tm
测试的训练数据集。在235名患者中,纳入“无癌症”组的患者(89名患者)是从因与泌尿科肿瘤学无关的问题而在泌尿科诊所就诊的年龄匹配的男性(n=58)以及接受过一次或多次12针诊断核心针活检而显示无前列腺癌证据(nepc)
的男性(n=30)中精心挑选的。
[0187]“癌症”群组中的患者(n=146)是根据核心针活检的组织病理学选择的。在146“癌症”群组中,90名患者被归类为gg1癌症,56名患者被归类为gg2
‑
5。
[0188]
在使用专有选择算法询问以分离结果“信息性的”与没有信息性的那些的来自训练数据集的6,599个微阵列序列中,只有400
‑
600个是信息性的。结果是指当添加每个序列以预测未知疾病状态的受试者是否患有前列腺癌和癌症分期(惰性与侵袭性)时影响算法的sncrna序列。
[0189]
使用的统计分析基于识别与结果有隐藏关联的序列的能力,这些关联仅在对其他序列进行调节后才能观察到。在400
‑
600个信息性的sncrna序列中,280个sncrna序列被用于分类算法,作为定义发现pca测试(图5)、发现hg测试(图7)和发现cs测试(图6)的表达特征的基础。然后为每个测试确定被认为具有最高重要性的信息性sncrna的子集(分别地图5、7和6(右小图))。
[0190]
将这280个sncrna结合以设计openarray
tm
平台,该平台为sentinel
tm
pca和cs测试提供了基础。sentinel
tm
pca测试结合了84个独特sncrna的聚集表达谱:60个mirna和24个snorna,用于将疾病状态未知的受试者分类为患有前列腺癌或没有前列腺癌。类似地,sentinel
tm
cs测试利用135个独特的sncrna:105个mirna和30个snorna,以将患有前列腺癌的受试者分类为患有gg1(惰性)前列腺癌或gg2
‑
gg5(侵袭性)前列腺癌。此外,61个sncrna(25个mirna和36个snorna)在两个测试中都提供了信息。openarray
tm
平台依次询问从尿液外来体中提取的单个sncrna样品中存在的信息性rna实体,而不会影响两项测试的灵敏度和特异性。
[0191]
实施例2
[0192]
在病例对照患者群组中验证sentinel
tm pca、sentine1
tm
cs和sentinel
tm
hg测试
[0193]
使用openarray
tm
平台的sentinel
tm
pca和sentinel
tm
cs测试的性能特征是在1436名患者的病例对照研究中建立的(表5)。
[0194]
表5:用于验证sentinel
tm
pca和sentinel
tm
hg测试的病例对照样品的人口统计学和临床特征。
[0195][0196]
*在豁免研究状态下,没有癌症证据的个体患者无法获得psa水平,所有患者均低于3.0ng/ml。
[0197]
sentinel
tm
pca测试的性能特征是在600名男性的病例对照群组中确定的,其人口统计数据如表5所示。sentinel
tm
pca分数的散点图如图8a所示,对应的接收者操作曲线(roc)曲线在图8c中。如表6中总结的,sentinel
tm
pca测试正确地将281/300名患者分类为患有癌症,将275/300名患者分类为没有癌症(灵敏度93.7%,特异性91.7%)。
[0198]
在600名男性的测试群组中确定了sentinel
tm
cs测试的性能特征。sentinel
tm
cs分数的散点图如图9a所示,对应的接收者操作曲线(roc)曲线在图9c中。如表6中总结的,sentinel
tm
cs测试正确地将143/154名患者分类为高等级(gg3
‑
gg5),将132/143正确分类为非高等级(灵敏度92.9%,特异性90.4%)。
[0199]
在600名男性的测试群组中确定了sentinel
tm
hg测试的性能特征。sentinel
tm
hg分数的散点图如图10a所示,对应的接收者操作曲线(roc)曲线在图10c中。如表6中总结的,sentinel
tm
cs测试正确地将94/100名患者分类为高等级(gg3
‑
gg5),将191/200正确分类为非高等级(gg1 gg2)(灵敏度94%,特异性95.5%)。
[0200]
表6:sentinel
tm
pca、sentinel
tm
cs和sentinel
tm
hg测试的经验灵敏度、特异性、ppv
和npv
[0201]
sentinel
tm
pca
ꢀꢀꢀꢀꢀ1‑
错误率分子分母比例95%下限ci95%上限ci灵敏度2813000.9370.9050.960特异性2753000.9170.8820.944ppv2813060.9180.8840.945npv2752940.9350.9030.959sentinel
tm
cs
ꢀꢀꢀꢀꢀ1‑
错误率分子分母比例95%下限ci95%上限ci灵敏度1431540.9290.8800.962特异性1321460.9040.8480.944ppv1431570.9110.8590.948npv1321430.9230.8710.959sentinel
tm
hg
ꢀꢀꢀꢀꢀ1‑
错误率分子分母比例95%下限ci95%上限ci灵敏度941000.9400.8800.975特异性1912000.9550.9190.978ppv941030.9130.8460.956npv1911970.9700.9380.987
[0202]
*
npv或阴性预测值是在测试结果为阴性后该个体不患有该特定疾病的概率。
[0203][0204]
**
检测的灵敏度是所有实际患有该疾病的人中检测呈阳性的人的比例。
[0205][0206]
***
检测的特异性是所有实际未患该病的人中检测为阴性的人的比例。
[0207][0208]
****
ppv或阳性预测值是在检测结果呈阳性后该个体真正患有特定疾病的概率。
[0209][0210]
实施例3
[0211]
使用sceintific sentinel
tm
平台识别临床上无意义的pca的安全性和科学有效性研究。
[0212]
临床研究目的旨在验证scientific sentinel
tm
pca测试和scientific sentinel
tm
cs测试的性能特征,以(1)识别50
‑
80岁男性中的前列腺癌患者,对怀疑有前列腺癌进行针刺活检,和(2)区分50
‑
80岁有临床意义的前列腺癌(等级2或以上)男性与临床无意义的前列腺癌(等级组1)的那些。这些分类将与核心针活检和根治性前列腺切除术(如果可用)的结果进行比较。将确定灵敏度、特异性、阳性和阴性预测值。本研究是一项前瞻性、
观察性和非干预性研究。知情参与者将在研究过程中提供两个或更多尿液样品,并同意与研究团队分享相关的匿名临床数据。
[0213]
年龄在50至80岁之间、对怀疑患有前列腺癌的进行核心针活检以及以其他方式符合纳入和排除标准的参与者将被登记并为sentinel
tm
pca/cs测试提供尿液样品。该研究将评估基于所公开方法的sentinel
tm
pca测试和sentinel
tm
cs测试的特性,其使用分类算法以识别未来的前列腺癌患者并将前列腺癌分类为临床有意义的或临床无意义的。
[0214]
从核心针活检的结果进行癌症的“金标准”评估:没有阳性核心的参与者将被指定为“无癌症”;如果患有癌症的所有核心的组织病理学均不大于等级组1,则一个或多个核心中患有癌症的参与者将被指定为“无临床意义”前列腺癌;如果任何核心具有等级组2
‑
5,则参与者将被指定为患有“临床有意义”前列腺癌。
[0215]
每个登记的研究参与者将被跟踪一年。参与者将在每次访问期间提供尿液样品,以及将获得所有相关的临床数据,包括再活检、psa结果和根治性前列腺切除术(如果作为临床护理的一部分施用)的病理报告。随访结果(如果有)将用于结果分析。对于提供的每个尿液样品,将确定sentinel
tm
pca和cs测试并与可用的1年随访结果数据进行比较,以告知测试的灵敏度、特异性、阳性和阴性预测值。
[0216]
分类算法通过将灵敏度控制在或高于预先指定的水平(表示为1
‑
α)来使用函数;例如,此设计中假定的值为α=0.05,因此群体中的灵敏度至少为95%。注意,α的值表示测试的假阴性率,即对于真阳性的患者,测试为(错误地)阴性。
[0217]
为了描述如何计算sentinel
tm
pca分数的截止值以控制灵敏度,对于训练数据集中的每个参与者,将使用训练数据集的其余成员和仅他的小非编码rna(sncrna)序列计算sentinel
tm
pca分数;也就是说,训练数据集中每个患者的真实疾病状态将被隐藏,从而模仿未来患者分类的设置。然后计算sentinel
tm
pca测试中使用的截止值,以便训练数据集中对患有前列腺癌的患者的经验灵敏度对应于为至少1
‑
α的未来患者的群体灵敏度提供上限一侧的95%置信区间。
[0218]
利用从训练数据集预先确定的sentinel
tm
pca分数的这种截止值,将根据本提议研究中累积的预期参与者数据计算灵敏度、特异性、阳性和阴性预测值的相应值,以及相应的95%置信区间上限,其中每个活检结果被隐藏,即,仅使用参与者的sncrna序列。注意,这些错误率是指疾病状态未知的未来患者的分类。
[0219]
本技术中引用的任何专利、专利申请出版物或科学出版物通过引用整体并入。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
