1.本发明属于药物分离纯化领域,具体为一种异射干苷元的制备方法。
背景技术:
2.异射干苷元,又名异鸢尾黄素(isotectorigenin,ψ
‑
tectorigenin),为射干苷元的同分异构体,化学名称为5,7,4
′‑
三羟基
‑8‑
甲氧基异黄酮。射干苷元、异射干苷元结构式如下:
[0003][0004]
异射干苷元属于类黄酮化合物,长期以来,类黄酮被认为具有抗炎,抗氧化,抗过敏,保肝,抗血栓,抗病毒和抗癌活性,但由于异射干苷元的研究报道很少,故其具体的药效作用还不明确,其在药学上的应用价值目前也未知。
[0005]
专利us20080311634a1公开了一种从鸢尾科植物的愈伤组织中分离异射干苷元的方法,该方法需要先培养愈伤组织,再通过醇提的方法将愈伤组织中富集的异射干苷元提取出。该方法复杂,且成本高,需要植物栽培领域与植物提取领域间协作才能制备得到高质量的异射干苷元,因此在工业生产中难以推广应用。
技术实现要素:
[0006]
为了解决异射干苷元药效作用不明确,以及至今无简便、实用、大量制备高纯度异射干苷元的方法的技术问题,本发明提供了一种异射干苷元的用途。
[0007]
本发明提供了异射干苷元在制备治疗抗肿瘤的药物中的用途。
[0008]
进一步地,所述药物为治疗结肠癌、肺癌和/或肝癌的药物。
[0009]
本发明还提供了一种异射干苷元的提取方法,它是由川射干药材或射干苷元为原料,加na2co3水溶液提取,分离、纯化、即得异射干苷元。
[0010]
进一步地,所述川射干药材与na2co3水溶液的质量体积比为0.5~2g:20ml;所述射干苷元与na2co3水溶液的质量体积比为0.5~2g:1000ml;所述川射干药材为川射干药材粗粉;所述na2co3水溶液的浓度为0.02%~0.08%w/v,g/ml,优选0.05%w/v,g/ml;所述提取为煎煮提取或回流提取,优选煎煮提取,时间4小时。
[0011]
进一步地,所述分离的方法为:取提取液过滤,滤液调ph值至6.5
‑
7.5,冷却,加氯仿萃取,取氯仿萃取液减压浓缩,干燥,得粗提物。
[0012]
更进一步地,所述提取液的原料为川射干药材时,冷却后还包括加与滤液等体积
的石油醚萃取;所述冷却至室温;所述氯仿与滤液等体积。
[0013]
进一步地,所述纯化的方法包括制备液相色谱纯化法、湿柱色谱纯化法或干柱色谱纯化法。
[0014]
更进一步地,所述制备液相色谱纯化法为:
[0015]
粗提物溶解于甲醇,注入制备色谱柱,再用流动相甲醇
‑
水洗脱,收集响应值最大时段的洗脱液,减压浓缩,干燥得异射干苷元;
[0016]
更进一步地,所述粗提物与甲醇的质量体积比为5~15mg:1ml;所述制备液相色谱的色谱柱填料为十八烷基硅烷键合硅胶,柱温为室温,检测波长为263nm;所述流动相甲醇
‑
水的体积比25:75,流速为10ml/min。
[0017]
更进一步地,所述湿柱色谱纯化法为:
[0018]
粗提物溶解于氯仿,加1倍量w/v,g/ml的80目硅胶拌匀,干燥,研细,上样于硅胶柱,再用流动相氯仿
‑
甲醇梯度洗脱,收集洗脱液,每5倍量v/w,ml/g于80目硅胶的洗脱液为1份,取第20
‑
25份洗脱液合并,减压浓缩,干燥得异射干苷元;
[0019]
所述硅胶柱是采用氯仿湿法装柱的200~300目硅胶柱,其直径5~6cm,柱高80cm;
[0020]
所述氯仿
‑
甲醇梯度洗脱程序为:依次用体积比20:1、10:1、6:1、3:1的氯仿
‑
甲醇溶液洗脱,每个体积比的氯仿
‑
甲醇溶液用量为80目硅胶的80倍量v/w,ml/g。
[0021]
更进一步地,所述干柱色谱纯化法为:
[0022]
粗提物溶解于氯仿,加1倍量w/v,g/ml的100目硅胶拌匀,干燥,研细,上样于硅胶柱,再用展开剂氯仿
‑
甲醇展开,取出硅胶,切割成20等份,合并距上样处第10~14份切割段,甲醇洗脱,减压浓缩,干燥得异射干苷元;
[0023]
所述硅胶柱是填有10~40μm薄层层析硅胶的中空玻璃柱,其直径6~7cm,柱高100cm;所述氯仿
‑
甲醇的体积比为10:1。
[0024]
本发明通过研究川射干模拟人肠道环境条件下(ph8.5左右)水煎煮试验的化学成分变化,发现了异射干苷、异射干苷元的存在,并用制备液相分离出异射干苷元并鉴定了其化学结构,进而确认射干苷元在弱碱水煎煮下可部分转化为异射干苷元。同时,通过药理实验证明,本发明制备得到的异射干苷元具有抗肿瘤活性,对hct116、a549、hepg2肿瘤细胞株活性优于射干苷元。
[0025]
本发明的异射干苷元分离纯化方法,通过碱液煎煮,氯仿萃取降低了后续异射干苷元的纯化难度,并以制备液相色谱、湿法柱层析和干法柱层析纯化使样品中的大极性及低极性的杂质充分分离,可在短时间内(48小时)制备克级纯度达99%%以上的异射干苷元,提高异射干苷元产品的纯度和收率,降低了成本,同时由于操作简便,适合工业化生产,具有广阔的市场应用前景。
[0026]
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
[0027]
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
[0028]
图1自制射干苷元色谱图
[0029]
图2实施例1异射干苷元色谱图(纯度97.33%)
[0030]
图3实施例2异射干苷元色谱图(纯度99.71%)
[0031]
图4实施例3异射干苷元色谱图(纯度99.63%)
[0032]
图5实施例4异射干苷元色谱图(纯度99.29%)
[0033]
图6川射干水煎煮样品溶液总离子流图
[0034]
图7川射干弱碱水煎煮样品溶液总离子流图
[0035]
图8射干苷元弱碱水煮lc
‑
ms总离子流图
[0036]
图9试验例2射干苷元弱碱水煮uplc色谱图
[0037]
图10试验例3射干苷元弱碱水煮uplc色谱图
[0038]
图11试验例4射干苷元弱碱水煮uplc色谱图
[0039]
图12试验例5射干苷元弱碱水煮hplc色谱图
[0040]
图13试验例5射干苷元弱碱水煮样品ms图
[0041]
图14异射干苷元红外光谱图
[0042]
图15异射干苷元氢谱图
[0043]
图16异射干苷元碳谱图
具体实施方式
[0044]
本发明具体实施方式中使用的原料、设备均为已知产品,通过购买市售产品获得,其中,液质联用仪为美国agilent rrlc
‑
6410 triple quadrupole,核磁共振波谱仪为bruker av
ꢀⅱ‑
600mhz,hplc色谱仪为美国agilent1100液相色谱仪,半制备液相色谱仪为美国agilent1200,uplc色谱仪为美国waters超高效液相色谱仪acquity,挂瓶式冷冻干燥机为北京博医康fd
‑
id
‑
80;
[0045]
川射干购于四川省中药饮片有限责任公司,射干苷元为自制,纯度100%,见图1),na2co3、盐酸、石油醚、氯仿(分析纯)等均购于成都科龙化工试剂厂。
[0046]
实施例1:川射干药材弱碱水煮,氯仿萃取,制备液相分离
[0047]
1、制备:
[0048]
取100g川射干粗粉,用0.05%na2co3水溶液20倍量(2l)煎煮2次,每次2小时,过滤,合并滤液,用1%hcl调ph至近中性,滤液浓缩至500ml,放冷至室温,依次用石油醚500ml、氯仿500ml萃取。取氯仿萃取液,60℃减压回收溶剂并干燥,即得含异射干苷元的提取物。取该提取物,用甲醇配制成10mg/ml浓度的溶液,用制备液相色谱仪纯化,制备柱填料为十八烷基硅烷键合硅胶,柱温为室温,流动相为甲醇:水(25:75),流速为10ml/min,检测波长263nm收集响应值最大时段的洗脱液,60℃减压回收溶剂,真空减压干燥,即得异射干苷元10.5mg。
[0049]
2、检测
[0050]
采用uplc检测(面积归一化法)异射干苷元,具体检测条件如下:
[0051]
waters acquity uplc系统;色谱柱为waters beh c18柱(2.1mm
×
50mm,1.7μm);流动相为乙腈
‑
水,梯度洗脱,0~14min(10:90~22:78),14~18min(22:78~37:63);体积
流量0.5ml.min
‑1;柱温:30℃;检测波长为263nm;进样量1μl。理论塔板数按异射干苷元计算均不得低于4000。
[0052]
3、结果
[0053]
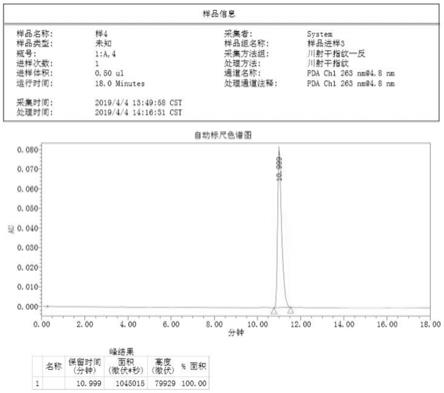
见图2,从图2可见异射干苷元纯度为97.33%。
[0054]
(备注:试验时间约72小时)
[0055]
实施例2:射干苷元弱碱水煮,氯仿萃取,制备液相分离
[0056]
1、制备
[0057]
取500mg射干苷元,用0.05%na2co3水溶液500ml煎煮4小时,煎煮液用1%hcl调ph至近中性,放冷至室温,用氯仿500ml萃取。取氯仿萃取液,60℃减压回收溶剂并干燥,即得含异射干苷元的提取物。取该提取物,用甲醇配制成10mg/ml浓度的溶液,用制备液相色谱仪纯化,制备柱填料为十八烷基硅烷键合硅胶,柱温为室温,流动相为甲醇:水(25:75),流速为10ml/min,收集响应值最大时段的洗脱液,60℃减压回收溶剂,真空减压干燥,即得异射干苷元75.4mg。
[0058]
2、检测
[0059]
采用uplc检测(面积归一化法)异射干苷元,具体检测条件同实施例1
[0060]
3、结果
[0061]
见图3,从图3可见异射干苷元纯度为99.71%。
[0062]
(备注:试验时间约72小时)
[0063]
实施例3:射干苷元弱碱水煮,氯仿萃取,湿柱层析分离
[0064]
1、制备
[0065]
取5g射干苷元,用0.05%na2co3水溶液5000ml煎煮4小时,煎煮液用1%hcl调ph至近中性,浓缩至1000ml,放冷至室温,用氯仿1000ml萃取。取氯仿萃取液,60℃减压回收溶剂并干燥,即得含异射干苷元的提取物。取该提取物,加氯仿10ml溶解,再用10g 80目硅胶拌匀吸附,干燥,研细,作为上柱样品。样品用200~300目硅胶柱层析,采用氯仿湿法装柱(中空玻璃柱,直径5~6cm,柱高80cm),氯仿
‑
甲醇梯度洗脱(20:1、10:1、6:1、3:1,v/v),每个体积比的氯仿
‑
甲醇溶液用量为800ml收集洗脱液(每50ml为一份),合并第20~25流份的洗脱液,60℃减压回收溶剂,真空减压干燥,即得异射干苷元0.851g。
[0066]
2、检测
[0067]
采用uplc检测(面积归一化法)异射干苷元,具体检测条件同实施例13、结果
[0068]
见图4,从图4可见异射干苷元纯度为99.63%。
[0069]
(备注:试验时间约96小时)
[0070]
实施例4:射干苷元弱碱水煮,氯仿萃取,干柱层析分离
[0071]
取5g射干苷元,用0.05%na2co3水溶液5000ml煎煮4小时,煎煮液用1%hcl调ph至近中性,浓缩至1000ml,放冷至室温,用氯仿1000ml萃取,取氯仿萃取液,60℃减压回收溶剂并干燥,即得含异射干苷元的提取物。取该提取物,加氯仿10ml溶解,再加10g粗硅胶(100目),拌匀,干燥,研细,作为上柱样品,样品用10~40μm薄层层析硅胶干柱层析(中空玻璃柱,直径6~7cm,柱高100cm),用展开剂氯仿
‑
甲醇100:5(v/v)展开,展开完毕,在通风柜中取出薄层层析硅胶,切割成20等份,合并距上样处第10~14份切割段,甲醇洗脱,60℃减压回收溶剂,真空减压干燥,即得异射干苷元0.947g。
[0072]
2、检测
[0073]
采用uplc检测(面积归一化法)异射干苷元,具体检测条件同实施例1
[0074]
3、结果
[0075]
见图5,从图5可见异射干苷元纯度为99.29%。
[0076]
(备注:试验时间约48小时)
[0077]
以下通过试验例进一步说明本发明的有益效果
[0078]
试验例1:川射干药材射干苷、射干苷元异构化研究
[0079]
1、水煎煮川射干药材
[0080]
称取川射干100g,粉碎,用20倍量水煎煮2次,每次2小时,过滤,合并滤液,减压浓缩至干。取25mg提取物粉末,用色谱甲醇溶解并定容至10ml量瓶中,过0.45μm微孔滤膜,即得样品溶液。
[0081]
对川射干水煎煮样品溶液进行lc
‑
ms检测
[0082]
检测条件:
[0083]
色谱柱:安捷伦c
18
柱,流动相:
[0084]
时间流速b相(有机相)00.330350.348450.370470.370
[0085]
流速:0.3ml/min;柱温:30℃;进样量:2μl。
[0086]
质谱条件:采用电喷雾离子化源(esi),正离子模式,喷雾电压4000v,源温度为100℃;雾化气为氮气,雾化压力为40psi;去溶剂气为氮气,温度300℃,流速为10l/min;碰撞气为高纯氮气,压力为0.1mpa;采用全扫描模式(m/z 50
‑
1000)对药物检测。
[0087]
总离子流图见图6,液质联用分析见表1.
[0088]
表1 川射干水煎煮样品溶液液质联用分析表
[0089]
序号出峰时间/min质荷比[m h]
质谱主要碎片化合物18.652463.20301射干苷29.467493.10331、118鸢尾甲苷b312.329493.20331鸢尾甲苷a413.148523.20361、118野鸢尾苷528.440301.10 射干苷元629.836331.10118鸢尾甲黄素b732.465331.10118鸢尾甲黄素a833.025361.10331、149、118野鸢尾黄素
[0090]
从表1可以看出,川射干中8个主要异黄酮类成分均被检出,结合参考文献,对其做了归属。
[0091]
2、碱水煎煮川射干药材
[0092]
称取川射干100g,粉碎,用20倍量0.05%na2co3水溶液煎煮2次,每次2小时,过滤,合并滤液,滤液用1%hcl调ph至近中性,减压浓缩至干。取25mg提取物粉末,用色谱甲醇溶
解并定容至10ml量瓶中,过0.45μm微孔滤膜,即得样品溶液。
[0093]
对川射干弱碱水煎煮样品溶液lc
‑
ms检测。
[0094]
检测条件:
[0095]
色谱柱:安捷伦c
18
柱。流动相:
[0096][0097]
流速:0.3ml/min;柱温:30℃;进样量:2μl。
[0098]
质谱条件:采用电喷雾离子化源(esi),正离子模式,喷雾电压4000v,源温度为100℃;雾化气为氮气,雾化压力为40psi;去溶剂气为氮气,温度300℃,流速为10l/min;碰撞气为高纯氮气,压力为0.1mpa;采用全扫描模式(m/z 50
‑
1000)对药物检测。
[0099]
总离子流图见图7,液质联用分析见表2.
[0100]
表2 川射干弱碱水煎煮样品溶液液质联用分析表
[0101]
[0102][0103]
表1与表2比较,川射干中8个主要异黄酮成分一一对应,但川射干弱碱水煎煮样品溶液多出2个峰(即峰2、峰10),质荷比与射干苷、射干苷元一致,故暂归属为异射干苷、异射干苷元。说明川射干在弱碱水煎煮条件下,射干苷、射干苷元可异构化。
[0104]
试验例2:射干苷元纯品的射干苷元异构化研究
①
[0105]
称取射干苷元纯品5mg于25ml圆底烧瓶中,加0.05%na2co3水溶液10ml,于水浴加热回流2小时,取出,溶液用1%hcl调ph至近中性。取溶液2ml,用色谱甲醇定容至10ml容量瓶中,过0.45μm微孔滤膜,即得样品溶液。对此溶液进行lc
‑
ms检测。
[0106]
检测条件:
[0107]
色谱柱:安捷伦c
18
柱。流动相:
[0108]
时间流速b相(有机相)00.310140.322180.337
[0109]
流速:0.3ml/min;柱温:30℃;进样量:2μl。
[0110]
质谱条件:采用电喷雾离子化源(esi),正离子模式,喷雾电压4000v,源温度为100℃;雾化气为氮气,雾化压力为40psi;去溶剂气为氮气,温度300℃,流速为10l/min;碰撞气为高纯氮气,压力为0.1mpa;采用全扫描模式(m/z 50
‑
1000)对药物检测。
[0111]
并进行uplc检测(检测条件:采用waters acquity uplc系统;色谱柱为waters beh c18柱(2.1mm
×
50mm,1.7μm);流动相为乙腈
‑
水,梯度洗脱,0~6min(10:90~40:60);体积流量0.4ml.min
‑1;柱温:30℃;检测波长为263nm;进样量1μl。理论塔板数按异射干苷元计算均不得低于4000。)
[0112]
lc
‑
ms总离子流图见图8,uplc见图9,lc
‑
ms样品分析见表3,uplc样品分析见表4。
[0113]
表3 射干苷元弱碱水煮样品lc
‑
ms分析表
[0114][0115]
表4 射干苷元弱碱水煮样品uplc分析表
[0116]
序号保留时间(min)峰面积(%)化合物
110.1458.85异射干苷元210.93891.15射干苷元
[0117]
从上图及表可以看出,射干苷元纯品在弱碱水煎煮条件下,发生异构化生成异射干苷元,2小时已有8.85%的分解。
[0118]
试验例3:射干苷元纯品的射干苷元异构化研究
②
[0119]
称取射干苷元纯品5mg于25ml圆底烧瓶中,加0.05%na2co3水溶液10ml,于水浴加热回流4小时,取出,溶液用1%hcl调ph至近中性。取溶液2ml,用色谱甲醇定容至10ml容量瓶中,过0.45μm微孔滤膜,即得样品溶液。对此溶液进行uplc检测(色谱条件见试验例2),色谱图见图10。
[0120]
从图10可以看出,射干苷纯品在0.05%na2co3水溶液中水浴加热回流4小时,异射干苷元峰面积为19.45%。
[0121]
试验例4:射干苷元纯品的射干苷元异构化研究
③
[0122]
称取射干苷元纯品5mg于25ml圆底烧瓶中,加0.05%na2co3水溶液10ml,于水浴加热回流6小时,取出,溶液用1%hcl调ph至近中性。取溶液2ml,用色谱甲醇定容至10ml容量瓶中,过0.45μm微孔滤膜,即得样品溶液。对此溶液进行uplc检测(色谱条件见试验例2),色谱图见图11。
[0123]
从图11可以看出,射干苷纯品在0.05%na2co3水溶液中水浴加热回流6小时,异射干苷元峰面积为20.80%。
[0124]
从试验例2、3、4结果可以看出,射干苷纯品弱碱回流提取2、4、6小时,异射干苷元分别为8.85%、19.45%及20.80%,当提取时间从4小时到6小时,异射干苷元升高幅度不大,故从成本考虑,确定提取4小时为宜;由于回流提取适宜小量样品提取,煎煮提取适宜大量样品的提取,考虑到实际工业化大生产,故最终确定分离异射干苷元样品由射干苷元弱碱水煎煮4小时。
[0125]
试验例5:异射干苷元结构测定初探
[0126]
称取射干苷元纯品5mg于25ml圆底烧瓶中,加0.1%na2co3水溶液10ml(ph9.0~10.0),于水浴加热回流4小时,取出,溶液用1%hcl调ph至近中性,加入氯仿萃取,取氯仿萃取液,60℃减压回收溶剂并干燥,得淡黄色干燥疏松粉末。用hplc检测,检测条件:色谱柱为kromasil c18柱(4.6mm
×
150mm,5μm),流动相为甲醇
–
水(30:70),流速1ml/min;柱温:30℃;检测波长为282nm;进样量10μl,纯度100%,并进行ms检测,结果见图12、图13。初步推测其结构为
[0127][0128]
从本试验例可以看出,射干苷元在0.1%na2co3水溶液(ph9.0~10.0)条件下水浴加热回流4小时,可生成另一化合物,而并非异射干苷元。
[0129]
试验例6:异射干苷元结构测定
[0130]
异射干苷元(实施例4样品),淡黄色针晶,盐酸镁粉反应呈玫瑰红色,三氯化铝反应呈鲜黄色,gibbs反应呈阴性,uvλ
max
263 nm,ir、1h nmr、
13
c nmr见图14~16,故鉴定为5,7,4
′‑
三羟基
‑8‑
甲氧基异黄酮,即异射干苷元,化学结构式如下:
[0131][0132]
试验例7:异射干苷元抗肿瘤活性
[0133]
1、试验目的
[0134]
研究射干苷元、异射干苷元体外抑制hct116、a549、hepg2、mcf7肿瘤细胞增殖作用。
[0135]
2、试验材料
[0136]
2.1细胞株
[0137]
hct116、a549、hepg2肿瘤细胞株购自中科院细胞保存库。以含有10%胎牛血清的rpmi
‑
1640培养基(含青霉素100u/ml,链霉素100μg/ml)培养于37℃、5%co2、饱和湿度细胞培养箱内。每隔2
‑
3d,以0.05%胰蛋白酶
‑
0.53mm edta消化传代。
[0138]
mcf7肿瘤细胞株购自中科院细胞保存库。以含有10%胎牛血清的rpmi
‑
1640培养基(含青霉素100u/ml,链霉素100μg/ml,重组人胰岛素0.4u/ml)培养于37℃、5%co2、饱和湿度细胞培养箱内。每隔4
‑
5d,以0.05%胰蛋白酶
‑
0.53mm edta消化传代。
[0139]
2.2样品
[0140]
试验样品来源于四川省中医药科学院中药药学研究所(实施例4样品)。以dmso溶解配制成100mg/ml溶液/混悬液,
‑
20℃保藏备用。
[0141]
2.3试剂
[0142]
胎牛血清购自bovogen公司,rpmi
‑
1640培养基、mtt、胰蛋白酶、edta、dmso购自sigma公司,其余试剂均为国产分析纯。
[0143]
3试验方法
[0144]
分别取对数生长期的肿瘤细胞开展试验,hct
‑
116、a549、hepg2细胞按3
×
103个细胞加入96孔板,mcf7细胞按8
×
103个细胞加入96孔板,每孔180μl,培养过夜。在96孔板中分别加入设计浓度的各样品20μl,培养72h。培养结束前4h,吸去培养液,加入earle’s bss100μl。加入mtt溶液(5mg/ml)10μl,孵育4h后,每孔加入10%sds溶液(0.01m hcl配制)100μl,置细胞培养箱中孵育过夜,采用酶标仪570nm处测定od值。计算各样品浓度的抑制率,采用curve expert计算ic
50
值。
[0145]
4试验结果
[0146]
各样品对hct
‑
116、a549、hepg2、mcf7细胞增殖抑制率和ic
50
如表5所示。
[0147][0148]
从上表可以看出,异射干苷元具有抗肿瘤活性,对3种人癌细胞,即结肠癌(hct116)、肺癌(a549)、肝癌(hepg2)均强于射干苷元,故其可作为抗肿瘤药物应用,或作为
先导化合物进行结构修饰研究等。
[0149]
综上,本发明的异射干苷元分离纯化方法,通过碱液煎煮,氯仿萃取降低了后续异射干苷元的纯化难度,并以制备液相色谱、湿法柱层析和干法柱层析纯化使样品中的大极性及低极性的杂质充分分离,可在短时间内制备克级纯度达99%%以上的异射干苷元,提高异射干苷元产品的纯度和收率,降低了成本,同时由于操作简便,适合工业化生产,具有广阔的市场应用前景。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。