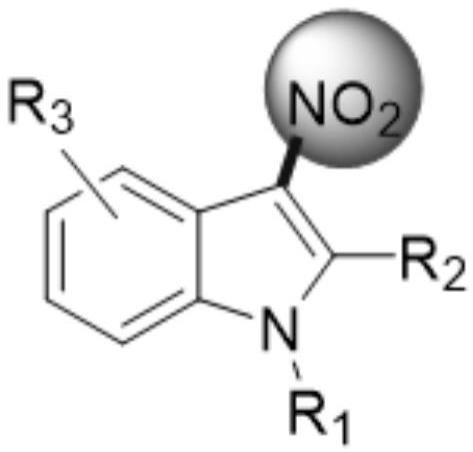
1.本发明涉及有机合成技术领域,具体而言,涉及一种3-硝基吲哚类似物及其制备方法。
背景技术:
2.3-硝基吲哚类似物是一种非常重要的多用途化工原料。其在抗肿瘤,抗菌,荧光材料以及天然产物核心骨架的构建中发挥着及其重要的作用。但由于其制备方法的缺陷限制了它的广泛应用。吲哚核是许多具有重要生物活性的天然分子和合成分子的重要组成部分。它是制药行业的“特权结构”之一,因为该片段在药物发现中起着核心作用。尽管已经研究了吲哚基序的富电子特性数十年,但最近开发利用3-硝基吲哚衍生物的亲电子反应性已成为许多具有挑战性的化学反应的核心。
3.目前,3-硝基吲哚类似物的合成虽然有文献报道,但依然存在一些不足之处,比如合成过程需要用到浓硝酸:反应收率过低,后期纯化较难;反应条件剧烈,存在爆炸风险;反应温度-78℃左右,能耗高;并且强酸腐蚀设备;大批量生产受限且安全风险较高。
技术实现要素:
4.本发明的目的在于提供一种3-硝基吲哚类似物及其制备方法,其以各类吲哚为起始原料制备目标产物,经一步亲电硝化反应高效、安全地制备3-硝基吲哚类似物。重要的是,本发明开发了一种不用硝酸即可在接近室温的条件下制备3-硝基吲哚类似物的合成方法,反应的机理如下:
[0005][0006]
发明人经过研究发现:四甲基硝酸铵与三氟乙酸酐复分解反应形成三氟乙酸硝酸酯(a),a是一个很强的亲电试剂,它能够与各种吲哚衍生物形成四元环状中间体b,b进一步转化形成各种3-硝基吲哚衍生物。弥补了当前制备该类化合物合成方法的缺陷及不足。本发明合成工艺设备简单,操作容易,对环境友好,成本低,收率高,可商业化大规模的制备和生产,满足当前不断增长的市场需求。
[0007]
本发明的实施例通过以下技术方案实现:
[0008][0009]
以各类吲哚为原料,加入溶剂、硝酸盐,在-20~25℃搅拌下滴入酸酐,在-20~25℃下反应1-8h,然后经萃取、干燥、过滤,减压蒸除溶剂得3-硝基吲哚类似物粗品,粗品进一步纯化得纯品。
[0010]
本发明实施例的技术方案至少具有如下优点和有益效果:
[0011]
本发明合成3-硝基吲哚衍生物的方法,不使用硝酸即可在接近室温的条件下高收率得到3-硝基吲哚类似物,且副产物少,所使用的原料价格低廉、易得,反应条件温和,对环境友好,成本低,收率高,可商业化大规模的制备和生产,满足当前不断增长的市场需求。
具体实施方式
[0012]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0013]
下面对本发明实施例提供的一种3-硝基吲哚类似物的制备方法进行具体说明。
[0014]
一种3-硝基吲哚类似物的制备方法,包括以下步骤:
[0015][0016]
以吲哚衍生物为原料,加入溶剂、硝酸盐,在一定反应温度下滴入酸酐反应一段时间后经萃取、干燥、过滤,减压蒸除溶剂得3-硝基吲哚类似物粗品,粗品进一步纯化得纯品3-硝基吲哚类似物。
[0017]
进一步地,所述硝酸盐与吲哚衍生物的摩尔比为1:1-4:1。
[0018]
进一步地,所述吲哚衍生物与酸酐的摩尔比为1:1-1:8。
[0019]
进一步地,所述反应温度为-20~25℃,反应时间1-8h。
[0020]
进一步地,所述硝酸盐选自盐硝酸钾、硝酸钠、四甲基硝酸铵或四丁基硝酸铵中的一种或几种。
[0021]
进一步地,所述溶剂选自四氢呋喃、1,4-二氧六环、乙腈、二氯甲烷、1,2-二氯乙烷、乙酸乙酯或二甲苯中的一种或几种。
[0022]
进一步地,所述酸酐选自三氟乙酸酐、三氟甲磺酸酐或乙酸酐中的的一种或几种。
[0023]
进一步地,萃取时萃取溶剂为乙酸乙酯、二氯甲烷或1,2-二氯乙烷中的一种或几种。
[0024]
进一步地,所述化方法可采用柱色谱分离、重结晶中的一种或几种。
[0025]
以下提供几种具体的3-硝基吲哚类似物的制备方法。
[0026]
实施例1
[0027]
n-boc-2-甲基-3-硝基吲哚的制备:反应釜中依次加入23.1g n-boc-2-甲基吲哚、
15g四甲基硝酸铵,200ml乙腈,降温至15℃,加入21g三氟乙酸酐,反应5h,然后加入水淬灭反应,用乙酸乙酯萃取、无水硫酸镁干燥、过滤,减压蒸除溶剂得3-硝基吲哚类似物粗品、粗品经柱色谱纯化得纯品24g,收率88%。
[0028]1h nmr(400mhz,cdcl3):δ8.29-8.21(m,1h),8.12-8.05(m,1h),7.47-7.36(m,2h),3.09(s,3h),1.75(s,9h).
[0029]
13c nmr(101mhz,cdcl3):δ149.2,142.3,133.6,131.4,125.8,125.1,121.7,120.4,115.0,86.7,28.1,15.1.
[0030]
hrms(esi-tof)m/z:[m na] calcd for c
14h16
n2o4na 299.1008;found 299.1009.
[0031]
实施例2
[0032]
1-苄基-3-硝基吲哚的制备:反应釜中依次加入20.6g n-苄基吲哚、34g四丁基硝酸铵,200ml乙酸乙酯,降温至10℃,加入25g三氟乙酸酐,反应3h,然后加入水淬灭反应,用乙酸乙酯萃取、无水硫酸镁干燥、过滤,减压蒸除溶剂得3-硝基吲哚类似物粗品、粗品加入2倍量的无水乙醇升温至溶解完全,降温析晶。减压干燥即得纯品20g,收率80%。
[0033]1h nmr(400mhz,cdcl3):δ8.33(m,1h),8.10(s,1h),7.45-7.33(m,6h),7.27-7.19(m,2h),5.38(s,2h).
[0034]
13
c nmr(101mhz,cdcl3):δ135.4,134.4,130.6,129.3,129.1,128.8,127.5,124.7,124.4,121.1,121.0,111.0,51.3.
[0035]
hrms(esi-tof)m/z:[m na] calcd for c
15h12
n2o2na 275.0796;found 275.0798.
[0036]
实施例3
[0037]
n-boc-2-苯基-3-硝基吲哚的制备:反应釜中依次加入29.2g n-boc-2-苯基吲哚、14g四甲基硝酸铵,200ml乙酸乙酯,降温至0℃,加入28g三氟乙酸酐,反应6h,然后加入水淬灭反应,用乙酸乙酯萃取、无水硫酸镁干燥、过滤,减压蒸除溶剂得n-boc-2-苯基-3-硝基吲哚粗品、粗品经柱色谱纯化得纯品31.8g,收率94%。
[0038]1h nmr(400mhz,cdcl3):δ8.38-8.30(m,1h),8.30-8.21(m,1h),7.56-7.48(m,5h),7.46(m,2h),1.25(s,9h).
[0039]
13
c nmr(101mhz,cdcl3):δ148.7,140.4,134.3,131.0,130.8,129.4,129.4,128.2,126.6,125.4,121.3,120.9,114.9,86.0,27.2.
[0040]
hrms(esi-tof)m/z:[m na] calcd for c
19h18
n2o4na 361.1164;found 361.1166.
[0041]
实施例4
[0042]
n-boc-5-溴-3-硝基吲哚的制备:反应釜中依次加入59g n-boc-5-溴-吲哚、32g四甲基硝酸铵,400ml二氯甲烷,降温至25℃,加入48g三氟乙酸酐,反应5h,然后加入水淬灭反应,用二氯甲烷萃取、无水硫酸镁干燥、过滤,减压蒸除溶剂得n-boc-5-溴-3-硝基吲哚粗品、粗品经柱色谱纯化得纯品60g,收率85%。
[0043]1h nmr(600mhz,cdcl3):δ8.51(s,1h),8.42(d,j=2.0hz,1h),8.11(d,j=8.9hz,1h),7.56(dd,j=8.9,2.0hz,1h),1.71(s,9h).
[0044]
13
c nmr(151mhz,cdcl3)δ147.8,133.0,131.4,129.7,128.5,123.3,122.9,119.3,117.0,87.3,28.0.
[0045]
hrms(esi-tof)m/z:[m na] calcd for c
13h13
n2o4nabr 362.9956;found 362.9955.
[0046]
综上所述,本发明合成3-硝基吲哚衍生物的方法,不使用硝酸且可在接近室温的条件下高收率得到3-硝基吲哚类似物,且副产物少,所使用的原料价格低廉、易得,反应条件温和,对环境友好,成本低,收率高,可商业化大规模的制备和生产,满足当前不断增长的市场需求。
[0047]
以上仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
