1.本技术属于有机合成技术领域,尤其涉及一种分子骨架的合成方法。
背景技术:
2.近年来随着阿尔茨海默病、帕金森氏病等神经退化性疾病患者的增多,寻找一种能够诱导神经细胞生长从而达到修复神经损伤的药物成为了化学生物学家的研究热点。具有诱导神经元生长的天然产物debenzoyltashironin成为了焦点。
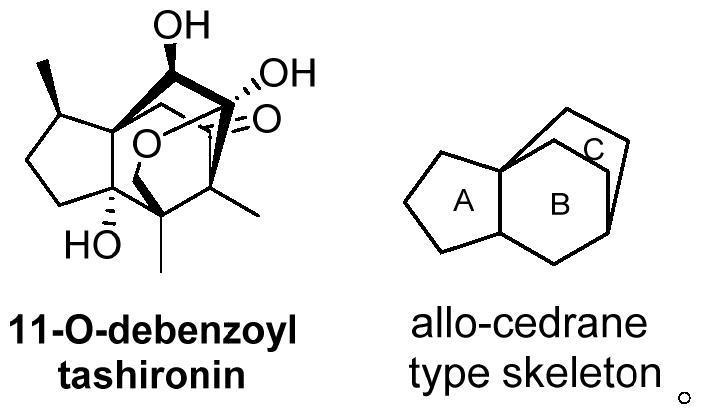
3.天然产物debenzoyltashironin是2001年日本德岛文理大学的fukuyama小组从滇缅八角(illicium merrillianum)的果皮中分离得到的倍半萜类化合物。它不仅拥有非常独特的生物活性,可以促进胎鼠大脑皮层神经元神经突起的增长,而且结构新颖,具有bicyclo[2.2.2]octanes双环体系,且拥有半缩酮氧桥环,高度的官能团化以及复杂的笼状结构使其成为一个极具合成挑战的分子。关于天然产物debenzoyltashironin的全合成报道较少,更多的是围绕该分子的骨架即allo-cedrane型骨架的合成。debenzoyltashironin以及allo-cedrane型骨架的化学结构式如下所示:
[0004][0005]
目前,关于该天然产物debenzoyltashironin及其骨架的合成路线较长,条件苛刻,构建效率较低,合成成本高,因此制约了其进一步研究。
技术实现要素:
[0006]
本技术的目的在于提供一种天然产物骨架的合成方法,旨在解决如何简单低成本地合成天然产物debenzoyltashironin的骨架的技术问题。
[0007]
为实现上述申请目的,本技术采用的技术方案如下:
[0008]
本技术提供一种分子骨架的合成方法,包括如下步骤:
[0009]
将三元环化合物1和卤化锌进行开环反应得到锌试剂2;
[0010]
将所述锌试剂2转化成铜试剂后与不饱和炔化合物3进行环加成反应得到亲双烯体化合物4;
[0011]
将所述亲双烯体化合物4与烯醇硅醚化合物5进行diels-alder反应得到五六并环化合物6;
[0012]
将所述五六并环化合物6脱除保护基得到化合物7;
[0013]
将所述化合物7进行氧化反应到醛基化合物8;
[0014]
将所述醛基化合物8进行羟醛反应得到分子骨架9;
也旨在包括多数形式,除非上下文清楚地表示其他含义。
[0023]
本技术实施例说明书中所提到的相关成分的重量不仅仅可以指代各组分的具体含量,也可以表示各组分间重量的比例关系,因此,只要是按照本技术实施例说明书相关组分的含量按比例放大或缩小均在本技术实施例说明书公开的范围之内。具体地,本技术实施例说明书中所述的质量可以是μg、mg、g、kg等化工领域公知的质量单位。
[0024]
本技术实施例提供一种分子骨架的合成方法,包括如下步骤:
[0025]
s01:将三元环化合物1和卤化锌进行开环反应得到锌试剂2;
[0026]
s02:将锌试剂2转化成铜试剂后与不饱和炔化合物3进行环加成反应得到亲双烯体化合物4;
[0027]
s03:将亲双烯体化合物4与烯醇硅醚化合物5进行diels-alder反应得到五六并环化合物6;
[0028]
s04:将五六并环化合物6脱除保护基得到化合物7;
[0029]
s05:将化合物7进行氧化反应到醛基化合物8;
[0030]
s06:将醛基化合物8进行羟醛反应得到分子骨架9;
[0031]
其中,上述化合物的结构式如下:
[0032][0033]
本技术实施例提供的分子骨架的合成方法,是一种关于allo-cedrane型骨架的方法,该合成方法的路线设计简单,操作工艺简便,反应条件温和,线性步骤少,而且原料廉价易得,产物收率高,是一种构建allo-cedrane型骨架的独特方案,为具有该类骨架的分子全合成提供了新的思路与策略。
[0034]
具体地,本技术实施例的合成方法中,合成的该分子骨架可以是天然产物debenzoyltashironin相关的allo-cedrane型骨架,因此,本技术实施例的合成方法为天然
产物debenzoyltashironin全合成提供了新的思路与策略。
[0035]
步骤s01:三元环化合物1进行开环反应生成锌试剂2步骤。
[0036]
在一实施例中,将三元环化合物1和卤化锌进行开环反应的步骤包括:将三元环化合物1和卤化锌溶于醚类溶剂中,在超声条件下进行反应。该条件下,三元环化合物1可以很好地进行开环反应,使三元环化合物1(结构式中,et表示乙基,tms表示三甲基硅基团)打开三元环生成锌试剂2。
[0037]
具体地,卤化锌可以是氯化锌、溴化锌、碘化锌等试剂,具体实施例中使用氯化锌。而醚类溶剂可以是甲醚、乙醚、丙醚等有机溶剂,具体实施例中可以使用乙醚。
[0038]
具体地,超声条件下进行反应的条件包括:温度为20~30℃,时间为1~3h。该条件下可以进行充分反应生成锌试剂2。
[0039]
具体实施例中,具体操作包括:在氮气保护下,将卤化锌和醚类溶剂加入反应瓶中搅拌后,然后滴加三元环化合物1的醚类溶液,转移至超声仪上进行上述超声操作。
[0040]
步骤s02:亲双烯体化合物4生成步骤。
[0041]
在一实施例中,将锌试剂2转化成铜试剂后与不饱和炔化合物3进行环加成反应的步骤包括:将锌试剂2与溴化亚铜二甲硫醚混合搅拌,然后加入不饱和炔化合物3和六甲基磷酰胺进行[3 2]环加成反应。
[0042]
上述过程中,锌试剂2和溴化亚铜二甲硫醚(cubr
·
me2s)反应从而转化成铜试剂,该铜试剂然后与不饱和炔化合物3(结构式中,pmb表示对甲氧基苄基)和六甲基磷酰胺(hmpa)混合,进行[3 2]环加成反应生成亲双烯体化合物4。
[0043]
本技术实施例从商业可得的三元环化合物1出发,通过开环反应和formal[3 2]反应,从而可以在一个反应体系中可构建diels-alder反应前体即亲双烯体化合物4。
[0044]
在一实施例中,锌试剂2与溴化亚铜二甲硫醚混合搅拌的温度为0~5℃,时间为0.5~1h;该条件下可以很好地转化成铜试剂。
[0045]
在一实施例中,[3 2]环加成反应包括:先在0~5℃条件下反应0.5~1.5h,然后升温至20~30℃反应1.5~2.5h。具体地,在0~5℃的条件下将锌试剂2转化成铜试剂后,保持温度不变,与不饱和炔化合物3和六甲基磷酰胺(hmpa)混合,先在0~5℃条件下反应0.5~1.5h,然后升温至20~30℃反应1.5~2.5h。这样可以更充分地进行环加成反应得到亲双烯体化合物4。
[0046]
进一步地,[3 2]环加成反应结束后还包括用饱和氯化铵溶液进行淬灭。淬灭反应后,进行有机萃取可以得到亲双烯体化合物4。
[0047]
s03:五六并环化合物6生成步骤。
[0048]
在一实施例中,将亲双烯体化合物4与烯醇硅醚化合物5进行diels-alder反应的步骤包括:将溴化锌与亲双烯体化合物4与烯醇硅醚化合物5混合后,在20~30℃的条件反应0.5~1h。
[0049]
具体地,上述过程在溴化锌的催化下,使用分子筛,以二氯甲烷(dcm)为溶剂发生diels-alder反应,一步合成五六并环化合物6。具体地,将亲双烯体化合物4溶于二氯甲烷中,加入溴化锌和分子筛搅拌,然后加入烯醇硅醚化合物5(结构式中,tips为三异丙基硅基)在20~30℃的条件反应0.5~1h,后续可以用饱和碳酸氢钠溶液淬灭,结束反应。进一步地,淬灭反应后进行有机萃取可以得到五六并环化合物6,其中三异丙基硅基烯醇硅醚的稳
定性好,后续经脱保护基和氧化条件依然可以保留下来本。
[0050]
s04:化合物7生成步骤。
[0051]
在一实施例中,将五六并环化合物6脱除保护基的步骤包括:将五六并环化合物6和2,3-二氯-5,6-二氰对苯醌溶于二氯甲烷和水的混合溶剂中,在20~30℃的条件反应10~30min。利用2,3-二氯-5,6-二氰对苯醌(ddq),水和二氯甲烷的混合溶剂,五六并环化合物6可以在常温下脱除对甲氧基苄基保护基得到化合物7。
[0052]
具体地,混合溶剂中,二氯甲烷与水的体积比可以为4~6:1。
[0053]
s05:醛基化合物8生成步骤。
[0054]
在一实施例中,将化合物7进行氧化反应的步骤包括:将化合物7和重铬酸吡啶盐氧化剂溶于二氯甲烷溶剂中进行一级羟基氧化。本技术实施例,化合物7可以在中性的温和氧化剂重铬酸吡啶盐(pdc)条件下,以二氯甲烷为溶剂,室温下氧化一级羟基得到醛基化合物式8。
[0055]
s06:分子骨架9生成步骤。
[0056]
在一实施例中,将醛基化合物8进行羟醛反应的步骤包括:将醛基化合物8和分子筛溶于二氯甲烷中,然后加入路易斯酸催化分子内mukaiyama羟醛反应。本技术实施例中,通过路易斯酸可以催化分子内mukaiyama羟醛反应,从而可以构建bicyclo[2.2.2]octanes双环骨架体系。
[0057]
进一步地,在-70~-80℃的条件下加入路易斯酸;具体加入的路易斯酸可以包括三氟化硼乙醚(bf3·
et2o)。加入路易斯酸后,mukaiyama羟醛反应的温度为20~30℃,时间为16~20h。醛基化合物8发生分子内mukaiyama羟醛反应的具体步骤包括:在氮气保护下,加入分子筛,醛基化合物8溶于二氯甲烷中,在-70~-80℃的条件下滴加路易斯酸,升温至20~30℃,反应16~20h。加水淬灭,即可得到分子骨架9。
[0058]
本技术实施例提供的合成方法,具体合成路线简单合理,操作简便,反应条件温和,线性步骤少,产品收率高,原料和试剂廉价易得,从商业可得的简单三元环化合物1计算,产品总收率可以高达10%,能够大幅降低生产成本,为debenzoyltashironin家族骨架的合成奠定了基础。具体地,从商业可得的三元环化合物1出发,可在一个反应体系中进行开环反应以及formal[3 2]反应,从而构建diels-alder反应前体亲双烯体化合物4。之后与烯醇硅醚化合物5一步合成五六并环化合物6,而该分子结构中的三异丙基硅基烯醇硅醚的稳定性非常好,后续经过脱保护基、氧化反应的两步条件依然能够保留下来。因此缩短了反应步骤,降低生产成本。经过脱保护基和氧化处理后,生成的醛基化合物8利用路易斯酸催化分子内mukaiyama aldol反应构建bicyclo[2.2.2]octanes双环骨架体系,从而可以得到分子骨架9。本技术实施例开创性地利用分子内mukaiyama aldol反应的策略构建六元桥环,为后续桥环的构建提供了思路,奠定了一定的实践基础。
[0059]
下面结合具体实施例进行说明。
[0060]
实施例1
[0061]
分子骨架9的制备方法,包括如下步骤:
[0062]
步骤1、亲双烯体化合物4的合成,过程如下:
[0063][0064]
氮气保护下,向烘干的50ml的梨形瓶中加入氯化锌(3.13g,23.0mmol)以及干燥的乙醚(9ml),在搅拌下滴加三元环化合物1(2.3ml,11.5mmol)的乙醚溶液(10ml),滴加5分钟。之后将反应体系移至超声仪上,25℃下超声2小时,生成锌试剂2。冷却至0℃后,氮气保护下加入溴化亚铜二甲硫醚(0.14g,0.6mmol),搅拌半小时后加入不饱和炔化合物式3(1.25g,5.7mmol)和六甲基磷酰胺(2ml,11.5mmol),接着在此0℃温度下反应1小时,后升至室温(25℃),保持2小时。加入饱和氯化铵溶液(10ml)淬灭。
[0065]
反应结束后有机相采用乙醚(50ml x3)萃取,合并萃取到的有机相用饱和食盐水(30ml)洗涤后经无水硫酸钠干燥,过滤,浓缩,硅胶快速柱层析色谱分离得到黄色油状液体(1.0g,59%)即为亲双烯体化合物4。rf=0.6(硅胶柱层析,正己烷:乙酸乙酯=20:1);收率:50%。
[0066]
亲双烯体化合物4:1h nmr(400mhz,cdcl3)δ6.99
–
6.96(d,2h),6.91
–
6.84(d,2h),4.83(s,2h),3.82(s,3h),3.69(s,3h),3.35(t,2h),2.98
–
2.96(t,2h),2.08
–
2.06(m,2h),2.01
–
1.96(m,2h).
13
c nmr(100mhz,cdcl3)δ201.01,180.14,165.02,159.30,131.01,129.26,114.52,73.70,68.42,55.63,52.18,46.78,35.61,34.67,27.69.
[0067]
步骤2、五六并环化合物6的合成,过程如下:
[0068][0069]
将亲双烯体化合物4(0.15g,0.5mmol)加入到二氯甲烷(5ml)中,加入溴化锌(0.23g,1.0mmol),室温下搅拌5分钟,然后滴加烯醇硅醚化合物5(0.26g,1.2mmol),反应0.5小时,tlc检测原料反应完全。加入饱和碳酸氢钠溶液淬灭。
[0070]
反应结束后有机相采用二氯甲烷(10ml x3)萃取,合并萃取到的有机相用饱和食盐水(5ml)洗涤后经无水硫酸钠干燥,过滤,浓缩,硅胶快速柱层析色谱分离得到黄色油状液体(0.14g,55%),即为五六并环化合物6。rf=0.5(硅胶柱层析,正己烷:乙酸乙酯=15:1);收率:55%。
[0071]
五六并环化合物6:1h nmr(400mhz,cdcl3)δ7.30
–
7.21(m,2h),6.91
–
6.84(m,2h),4.58(s,1h),4.47
–
4.36(m,2h),3.81(s,3h),3.67(s,3h),3.62
–
3.52(m,2h),2.50
–
2.39(m,
1h),2.30
–
2.15(m,3h),2.02
–
1.79(m,5h),1.73
–
1.65(m,1h),1.13
–
0.98(m,18h).
13
c nmr(100mhz,cdcl3)δ215.66,171.53,159.31,152.39,130.56,129.37,113.92,108.10,73.00,66.86,62.37,55.40,52.28,46.78,38.85,36.05,34.64,25.74,22.79,18.09,18.06,12.69.
[0072]
步骤3、化合物7的合成,过程如下:
[0073][0074]
五六并环化合物6(46mg,0.08mmol)加入到二氯甲烷/水(1.5ml/0.3ml)的混合溶剂中,然后加入2,3-二氯-5,6-二氰对苯醌(30mg,0.13mmol),室温下搅拌15分钟,tlc检测原料反应完全。
[0075]
反应结束后加水稀释,然后有机相采用二氯甲烷(3mlx3)萃取,合并萃取到的有机相,碳酸氢钠饱和溶液(2ml)洗,用饱和食盐水(2ml)洗涤后经无水硫酸钠干燥,过滤,浓缩,硅胶快速柱层析色谱分离得到黄色油状液体(15.8mg,48%),即为化合物7。rf=0.3(硅胶柱层析,正己烷:乙酸乙酯=5:1);收率:48%。
[0076]
化合物7:1h nmr(400mhz,cdcl3)δ4.58(s,1h),3.85
–
3.74(m,2h),3.70(s,3h),2.45
–
2.32(m,2h),2.23
–
2.15(m,2h),1.98
–
1.90(m,2h),1.87
–
1.75(m,2h),1.71
–
1.61(m,2h),1.06
–
1.00(m,18h).
13
c nmr(100mhz,cdcl3)δ215.42,171.77,152.72,108.04,62.27,59.68,52.37,46.87,42.01,35.92,34.80,25.72,22.96,18.09,18.05,12.70.
[0077]
步骤4、醛基化合物8的合成,过程如下:
[0078][0079]
化合物7(15mg,0.04mmol)加入到二氯甲烷(0.5ml)中,加入重铬酸吡啶盐(15mg,0.04mmol)反应36小时。tlc检测原料反应完全。过滤,浓缩,硅胶快速柱层析色谱分离得到黄色油状液体(12.9mg,79%),即为醛基化合物8。rf=0.4(硅胶柱层析,正己烷:乙酸乙酯=20:1);收率:79%。
[0080]
醛基化合物8:1h nmr(400mhz,cdcl3)δ9.84(t,j=2.3hz,1h),4.76(s,1h),3.71(s,3h),2.65(dd,j=15.7,2.4hz,1h),2.57
–
2.43(m,1h),2.42
–
2.20(m,4h),2.09
–
1.93(m,4h),1.15
–
0.93(m,24h).
13
c nmr(100mhz,cdcl3)δ213.98,200.69,171.24,153.40,107.29,61.94,52.56,46.17,35.62,33.86,29.84,25.64,22.60,18.02,12.67.
[0081]
步骤5、分子骨架9的合成,过程如下:
[0082][0083]
醛基化合物8(100mg,0.245mmol)加入到干燥好的二氯甲烷(5ml)中,-78℃下滴加三氟化硼乙醚(0.5ml,0.49mmol)。之后升至室温,反应18小时。tlc检测原料反应完全。加入二氯甲烷(2ml)稀释,加水淬灭。
[0084]
将反应后的有机相采用二氯甲烷(5ml x3)萃取,合并萃取到的有机相用饱和食盐水(3ml)洗涤后经无水硫酸钠干燥,过滤,浓缩,硅胶快速柱层析色谱分离得到白色固体(18.5mg,30%),即为分子骨架9。rf=0.2(硅胶柱层析,正己烷:乙酸乙酯=3:1);收率:95%。
[0085]
分子骨架9:1h nmr(500mhz,cdcl3)δ5.50(dt,j=2.9,1.5hz,1h),5.46(d,j=2.1hz,1h),5.26(d,j=2.5hz,1h),4.31(dt,j=6.0,2.0hz,1h),2.58
–
2.49(m,1h),2.46
–
2.40(m,1h),2.37(d,j=19.0hz,1h),2.14(dd,j=19.0,1.4hz,1h),2.01
–
1.92(m,1h),1.89(dd,j=13.3,9.7hz,1h),1.79(d,j=6.4hz,1h),1.75(dt,j=2.8,1.5hz,3h),1.14(s,3h).
13
c nmr(125mhz,cdcl3)δ211.30,151.34,141.88,128.11,113.14,72.21,55.28,51.74,39.96,38.53,35.59,34.78,16.36,13.11.
[0086]
以上所述仅为本技术的较佳实施例而已,并不用以限制本技术,凡在本技术的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本技术的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。