glp1r激动剂nmdar拮抗剂缀合物
技术领域
1.本发明总体上涉及治疗性缀合物领域,更特别地涉及具有胰高血糖素样肽1(glp-1)受体活性和n-甲基-d-天冬氨酸受体(nmdar)拮抗剂的缀合物。
背景技术:
2.肥胖症是富足社会中人类和家畜(比如狗和猫)最普遍的营养病,远远超过了营养缺乏病的数量。作为减肥手术的替代方案,已经有许多人尝试生产减重药物来治疗肥胖症。这已经产生了通过在肠道中充当脂肪酶抑制剂来阻止脂肪的吸收,或者通过下丘脑中选择性血清素受体2c激动剂来抑制食物摄入而起作用的药物。
3.胰高血糖素样肽1(glp-1)是衍生自胰高血糖素原肽的组织特异性翻译后加工的30或31个氨基酸长的肽激素。glp-1类似物的最新适应症是用于减重,因为它作用于大脑的食欲调节中心。glp-1与食欲和体重维持相关,因为它对胃肠道有作用,对参与食欲调节的cns也有影响。它还可以延缓人的胃排空和肠道运动,这可能有助于调节食物摄入。现有技术已知用于治疗代谢性疾病的基于glp-1的疗法。parlevliet等人(j pharmacol exp ther.[药理学与实验治疗学杂志]2009年1月;328(1):240-8)
″
和相关专利申请ep 1968645 a2、ep 2125003 a2和ep 1843788 a2描述了包含glp-1肽的人glp-1 mimetibody
tm
用于治疗肥胖症和肥胖症相关障碍的用途。更特别地,parlevliet等人(2009)描述了特定的glp-1 cnto 736,它可以减少高脂肪喂养小鼠的食物摄入和体重(归因于脂肪量的降低)。
[0004]
nmdar拮抗剂通过抑制nmda受体的作用而起作用,并且有一些临床前证据表明,nmdar拮抗作用可能与食欲降低和体重维持相关。deng等人(2019,frontiers in psychiatry[精神病学前沿],10,第15篇)描述了盐酸美金刚(nmdar拮抗剂)在饮食诱导肥胖小鼠(由高脂肪饮食诱导)中的减重作用。smith等人(neuropsychopharmacology[神经心理药理学](2015)40,1163-1171)描述了美金刚可以选择性地在接受高适口、高糖饮食的大鼠中剂量依赖性地减少暴饮暴食,并完全阻断觅食行为和强迫性暴食。此外,popik等人(amino acids[氨基酸](2011)40:477-485)描述了在大鼠中长期施用盐酸美金刚可以选择性地减少高适口食物的摄入,对标准饮食的食用作用较小,并且这种作用在停止治疗后持续存在。
[0005]
美金刚治疗人类暴食症的作用也有报道。hermanussen和tresguerres(economics and human biology 3[经济学与人类生物学3](2005)329-337)报道了一项针对五名肥胖年轻女性的治疗试验,表明美金刚治疗可在最初24小时内显著减少食欲,抑制暴食症,并在几天内导致体重减少。brennan等人(int j eat disord[国际进食障碍杂志]2008;41:520-526)描述了一项初步研究,表明每天施用美金刚持续12周可改善人类受试者的暴饮暴食。
[0006]
人们越来越需要具有更大疗效、高安全性(低毒性作用)的新型减重治疗,这也提供了方便和安全的施用选择。
技术实现要素:
[0007]
鉴于上述情况,因此本发明的目的是提供一种有效且安全的治疗剂,以降低肥胖人类受试者的食物摄入和减轻体重。
[0008]
因此,本发明的第一方面涉及一种包含在glp-1受体处显示天然glp-1的至少0.1%活性的肽和n-甲基-d-天冬氨酸受体(nmdar)拮抗剂的缀合分子,该肽直接或通过化学接头共价键合到nmdar拮抗剂上。
[0009]
诸位发明人令人惊讶地发现,肽与glp-1受体激动作用和nmdar拮抗作用的缀合代表了有效逆转肥胖症的新的药物策略。如图3至13所示,与单独的glp-1肽、美金刚或mk801相比,基于该策略的缀合物在抑制食物摄入方面具有优势。此外,已经表明基于glp-1肽变体的缀合物,例如glp-1/抑胃多肽(gip)肽(seq id no:9)和可选的nmdar拮抗剂对食物摄入和体重降低具有类似的有益作用。这得到了测试glp-1/gip协同激动剂和nmdar拮抗剂奈拉美生(neramexane)的进一步发现的支持,分别如图33至34和图36至38所示。此外,虽然该缀合物得益于nmdar拮抗作用对减重的影响,但该策略避免了nmdar拮抗作用的中枢神经系统效应。在不受任何特定理论约束的情况下,诸位发明人推测,由于肽对glp-1受体的亲和力,nmdar拮抗剂通过在体内glp-1受体位点处和/或其附近聚集而实现了这种作用。
[0010]
肽将具有氨基末端和羧基末端。在本发明的上下文中,该氨基末端和该羧基末端也可分别称为n末端和c末端,以及相应的衍生形式。
[0011]
该肽可由遗传密码编码的氨基酸组成,或者它可含有由遗传密码编码的氨基酸和由非遗传密码编码的天然氨基酸,比如羟脯氨酸、γ-羧基谷氨酸、鸟氨酸、磷酸丝氨酸、d-丙氨酸(dala)和d-谷氨酰胺。此外,该肽可掺入合成氨基酸,比如d-丙氨酸和d-亮氨酸,或a-氨基异丁酸(aib)、d-丝氨酸(dser)、n-甲基丝氨酸。
[0012]
在优选实施方案中,该肽中第2位(从n末端开始计数)的氨基酸是dser、dala、aib、甘氨酸、n-甲基-ser或缬氨酸。
[0013]
该肽还可具有一个或多个修饰以稳定二级结构,比如在该肽第15位的谷氨酸和第20位的赖氨酸之间的环化,这些位置从n末端开始计数。
[0014]
该肽可从任何来源获得,或者该肽可以根据需要生产。例如,该肽可从组织中分离,或者该肽可通过本领域技术人员熟知的方法重组生产或合成。
[0015]
该缀合分子包含肽,该肽(以其游离形式)在该glp-1受体处显示天然glp-1的至少0.1%活性。在本发明的上下文中,glp-1受体活性,也可称为glp-1激活(glp-1r活性),可通过在体外测定中测量过量表达该glp-1受体的hek293细胞中的camp诱导进行测量。特别地,可以使用用编码该glp-1受体的dna和与camp应答元件连接的萤光素酶基因共转染的hek293细胞(报告分子测定)。该测定可如bech等人(j.med.chem.[药物化学杂志]2017,60,17,7434-7446)所述进行。使用该测定,可以测定来自每个缀合物的glp-1r活性,并相对于同一测定中天然glp-1(seq id no:1)肽所获得的活性进行呈现。在一个实施方案中,该缀合物的肽显示天然glp-1的至少1%活性,比如至少5%、10%、15%、20%或30%活性。
[0016]
nmdar拮抗剂将与nmdar结合,且nmdar拮抗剂可被描述为具有与特定nmda受体的解离常数kd,例如以nmdar拮抗剂的游离形式。nmdar拮抗剂通常具有纳摩尔范围内的解离常数,例如,mk801与不同物种的nmda受体的解离常数为在大鼠脑膜中kd=6.3nm,在小鼠脑匀浆中kd=10nm,而在猪脑中kd1.3nm。解离常数的确定对本领域技术人员而言是公知的。在
[0022]
该缀合分子的肽将具有足以使该肽(以其游离形式)在该glp-1受体处显示天然glp-1的至少0.1%活性的长度。通常来说,对于包含至少10个氨基酸的肽可以观察到这一点,但当该肽包含超过60个氨基酸时,可能无法显示出该活性。因此,在一个实施方案中,该肽的长度在10至60个氨基酸的范围内,例如20至50个氨基酸。与其他肽序列有一定百分比同一的本发明氨基酸序列应包含肽的足够的氨基酸序列,例如至少10个氨基酸,以通过本领域技术人员手动评估序列,或通过使用比如blast(基本局部比对搜索工具)等算法进行计算机自动序列比较和识别,提供对该肽的推定识别(综述参见altschul,等人,meth enzymol.[酶学方法]266:460,1996;和altschul,等人,nature genet.[自然遗传学]6:119,1994)。
[0023]
在本发明的上下文中,肽可通过具有天然或合成氨基酸的取代、插入和/或具有氨基酸缺失而在%同一性上不同。在一个实施方案中,其中该缀合物的肽具有seq id no:1的氨基酸序列。
[0024]
在一个实施方案中,通过乙酰化、脂肪酸缀合、二酸缀合、白蛋白缀合、小分子白蛋白结合剂和/或peg缀合来修饰该肽。还考虑了通过连接到比如抗体等载体蛋白而修饰的肽。修饰优选在该肽的第16、17、20、21、24、29、40位(从n末端开始计数)处、在c末端区内或在c末端氨基酸处。缀合可以通过任何合适的接头进行,比如通过二硫化物、马来酰亚胺、α-酮或基于点击化学的缀合。本领域技术人员知道如何制备此类缀合物。优选地,peg分子可以大于1kda,脂肪酸和二元酸可以含有多于12个碳原子。通常优选在修饰(peg/脂肪酸/二酸)和该肽之间添加间隔物,该接头优选为γ-glu接头,即短peg链。
[0025]
该缀合分子包含nmdar拮抗剂。任何nmdar拮抗剂均可与该缀合物一起使用。然而,优选nmdar拮抗剂是小分子,例如高达900kda。例如,在一个实施方案中,nmdar拮抗剂选自mk801、美金刚、氯胺酮、苯环利定(pcp)、奈拉美生和金刚烷胺。mk801、奈拉美生和美金刚是优选的。mk801和美金刚也是优选的。奈拉美生是与美金刚有关的化合物的非限制性实例,奈拉美生的作用如图36至38所示。
[0026]
本发明的肽和nmdar拮抗剂是共价键合的。在本发明的上下文中,该缀合分子也可称为肽-药物缀合物(pdc)。该肽和nmdar拮抗剂可以直接彼此键合。例如,nmdar拮抗剂可以通过酰胺键共价键合,该酰胺键是从nmdar拮抗剂上的氨基基团到该肽上的羧酸基团。这种酰胺键可以形成于具有羧酸基团比如谷氨酸残基、天冬氨酸残基、具有羧基的合成残基或c末端的羧酸的肽上的任何残基上。例如,当nmdar拮抗剂是mk801时,mk801的胺可以与该肽的氨基酸残基的羧酸结合。相应地,当nmdar拮抗剂是美金刚时,美金刚的胺可以与该肽的氨基酸残基的羧酸结合。
[0027]
在本发明的上下文中,直接共价键合意味着该肽与nmdar拮抗剂具有共价键,例如在两个分子之间不存在附加化学基团,比如接头基团。该肽和nmdar拮抗剂也可以通过化学接头键合。可以使用任何化学接头。然而,通常优选的是,该化学接头具有多达30个原子的长度。较长的链可具有使nmdar拮抗剂与该肽隔开的优点,使得当该肽与glp-1受体相互作用时,nmdar拮抗剂对该肽没有或几乎没有空间位阻。该肽的无空间位阻或低空间位阻提供对glp-1受体的更大亲和力。对glp-1受体具有更大亲和力的缀合物可能在glp-1受体位点处具有更大的聚集。化学接头优选为可裂解接头,比如酸可裂解接头、酶可裂解接头、肽可裂解接头或二硫化物接头,其在肽-药物缀合物中的用途在本领域中通常是熟知的。此类可
裂解接头的实例是包括葡糖苷酸、β-半乳糖苷、二硫化物、腙的化合物和/或可被半乳糖苷酶、葡萄糖苷酸酶、焦磷酸酶、磷酸酶、芳基硫酸酯酶、蛋白酶或酯酶裂解的化合物。例如,接头可包含可被组织蛋白酶切割的肽,比如gflg。该接头可进一步包含4-氨基苯甲酸(pab),其可通过酰胺或氨基甲酸酯键共价键合到nmdar拮抗剂的氨基基团上。该接头优选以其游离形式(即天然形式)释放nmdar拮抗剂,这可以通过许多不同的接头化学比如本文披露的二硫化物接头来实现。这些接头化学和附加接头化学是本领域技术人员熟知的。
[0028]
在一个实施方案中,nmdar拮抗剂在该肽的c末端区处共价键合。在本发明的上下文中,c末端区可以是从c末端计数多达50%的氨基酸,比如从c末端计数多达40%、30%、25%、20%或10%的氨基酸。例如,seq id no:1的c末端区可以是氨基酸21至40、26至40或31至40(从n末端计数的数字)。因此,nmdar拮抗剂,例如美金刚或mk801,可以直接或通过接头键合到从c末端计数的10个氨基酸中的任何一个。例如,nmdar拮抗剂,例如美金刚或mk801,可以直接键合到距离c末端5个氨基酸内的氨基酸上。因此,nmdar拮抗剂在该肽的n末端几乎没有产生或没有产生空间位阻。由于n末端参与与glp-1受体的结合,n末端的无空间位阻或低空间位阻可提供对glp-1受体更大的亲和力。对glp-1受体具有更大亲和力的缀合物可能在glp-1受体位点处具有更大的聚集。还考虑了可以将多于一种nmdar拮抗剂键合到同一肽分子上。
[0029]
在另一个高度优选实施方案中,nmdar拮抗剂通过包含二硫化物基团的化学接头共价键合到该肽上。二硫化物基团允许nmdar拮抗剂在被化学还原时从该肽中释放。包含二硫化物基团的化学接头(也称为二硫化物接头)确保该肽和该缀合物的nmdar拮抗剂在全身循环期间保持长时间的缀合。该二硫化物接头的二硫化物基团可在还原环境(比如细胞内环境)中还原,导致该缀合物被裂解,使得该缀合物的肽部分与该缀合物的nmdar拮抗剂部分分离。还原可以通过与例如硫醇(比如谷胱甘肽)或还原酶(比如细胞内蛋白质二硫化物异构酶)的二硫化物交换进行。该化学接头可以选自本领域已知的通式为r
′‑
s-s-r
″
的化学接头,其中r
′
和r
″
基团可以彼此相同或不同。如图3所示,实验表明,肽和nmdar拮抗剂的缀合物(通过包含二硫化物基团的化学接头缀合)具有约0.5至13小时的人血浆裂解半衰期。有利地,由于该肽对glp-1受体的亲和力,该缀合物可在体内glp-1受体位点处和/或其附近聚集,且nmdar拮抗剂可在glp-1受体位点处和/或其附近释放。当不含该缀合物的肽部分时,nmdar拮抗剂可在位点特异性nmdar结合时具有适当作用。诸位发明人推测,该缀合物可在紧邻含有glp-1受体的细胞的细胞外环境中裂解,或者该缀合物可以被含有glp-1受体的细胞内化且在细胞的还原环境中裂解。
[0030]
在一个实施方案中,该缀合分子通过化学接头缀合,其中该化学接头具有式r
1-r
3-s-s-r
4-r
5-o-co-r2,其中r1是该肽,r2是该nmdar拮抗剂,r3是任选的且当存在时选自c(ch3)2、ch
2-ch2或ch2,其键合到该肽的侧链或该肽的主链的碳原子,r4是(ch2)n或c6h4,r5是任选的且当存在时选自c(ch3)2、ch
2-ch2或ch2,并且n是1、2或3。当该化学接头被还原时,该缀合物中释放的nmdar拮抗剂部分经历分子内环化,这导致nmdar拮抗剂释放成其游离形式,见图1b。
[0031]
在一个实施方案中,该化学接头具有式r
1-r
3-s-s-(ch2)
n-o-co-r2,其中r1是该肽,r2是该nmdar拮抗剂,r3是任选的且当存在时选自c(ch3)2、ch
2-ch2或ch2,其键合到该肽的侧链或该肽的主链的碳原子,并且n是1、2或3。
[0032]
在一个实施方案中,该化学接头具有式r
1-r
4-r
3-s-s-(ch2)
n-o-co-r2,其中r1是该肽,r2是该nmdar拮抗剂,r3是任选的且当存在时选自ch(ch3)2、ch
2-ch2或ch2,其键合到该肽的侧链或该肽的主链的碳原子,r4是任选的且当存在时选自ch(ch3)2、ch
2-ch2或ch2,其键合到该肽的侧链或该肽的主链的碳原子,并且n是1、2或3。
[0033]
在一个实施方案中,第二自由基键与本发明的肽的主链相连。
[0034]
在另一个实施方案中,第二自由基键与本发明的肽的侧链相连。
[0035]
在本发明的上下文中,当r1键合到该肽的主链上时,c(ch3)2(l-青霉胺)可称为pen,ch
2-ch2(l-同型半胱氨酸)可称为hcys,且ch2(l-半胱氨酸)可称作cys,见图1a。
[0036]
如本文所用,第一和第二自由基键用于说明本文披露的化学接头中存在至少两个游离键。
[0037]
本发明有助于设计和合成包含通过化学接头附加的肽和nmdar拮抗剂的缀合分子的文库。图1显示了可以如何设计此类缀合分子。如图1a所示,可以通过将nmdar拮抗剂(图1中的mk801)与肽化学键合来制备缀合物。本领域技术人员将理解,可以通过本文披露的方法和文献中报道的其他方法制备大量不同的化学接头,且这些化学接头可以用于根据本文披露的和已知技术中其他地方报道的方法附加肽和nmdar拮抗剂。
[0038]
诸位发明人令人惊讶地发现,本发明的肽可作为减重药物和靶向剂发挥双功能作用,从而允许将其他非特异性小分子(比如nmdar拮抗剂)定点选择性递送至大脑中控制摄食的区域,这些区域可以是但不限于下丘脑核、最后区、孤束核和腹侧被盖区。因此,本发明的缀合分子提供了在控制食物摄入的大脑区域选择性调节谷氨酸能信号传导的途径,同时避免其在整个大脑中自由信号传导。应当理解,本发明的肽的靶向性质也可以促进nmdar拮抗剂递送至其他部位,比如内分泌胰腺。
[0039]
本文披露的缀合分子提供了选择性,且还专注于靶向区域中的药物作用。由缀合分子实现的这种靶向允许改善治疗指数,即更低的最小有效浓度。此外,偶联允许在glp-1受体靶向药物的疗效中添加另一层代谢药物作用。nmdar的组织选择性靶向可用于管理摄食行为,且可在治疗停止后减少复发,这是由于在较低体重设定点下突触可塑性重新巩固。
[0040]
诸位发明人已经证明了本发明的缀合物对食欲、食物摄入和体重的令人惊讶的协同作用,且这与单独施用肽或该药物所获得的效果相比显著更大,见图4至14。本发明的缀合物的令人惊讶的协同作用进一步得到了图21至28和图33至34以及图36至38所示的发现的支持。
[0041]
诸位发明人已经进一步证明了本发明的缀合物对食物奖赏和饱腹感的令人惊讶的协同作用,且这与单独施用肽或该药物所获得的效果相比显著更大,见图31。
[0042]
此外,本发明缀合物的协同作用已被证明与糖尿病患者的治疗相关,见图32。
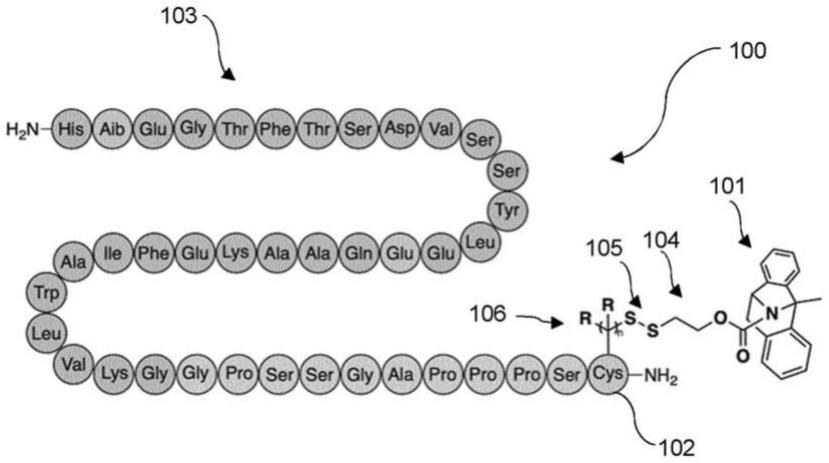
[0043]
因此,本发明的缀合物的施用导致肥胖动物的食物摄入和体重出乎意料地降低。
[0044]
在本发明的一个实施方案中,该缀合分子用于在疗法中使用。
[0045]
在一个实施方案中,本发明的缀合分子用于在治疗肥胖症、暴食症、胰岛素抵抗、2型糖尿病、血脂异常、非酒精性脂肪性肝炎或非酒精性脂肪性肝病中使用。
[0046]
本发明的另一方面涉及包含根据本发明的缀合分子和药学上可接受的载体的药物组合物。该缀合分子的任何实施方案都可用于该药物组合物中。
[0047]
在进一步的方面,本发明涉及根据本发明的缀合分子在制备药物组合物中的用
途。特别地,该药物组合物用于在治疗肥胖症、暴食症、胰岛素抵抗、2型糖尿病、血脂异常、非酒精性脂肪性肝炎或非酒精性脂肪性肝病中使用。该缀合分子的任何实施方案可用于制备该药物组合物。
[0048]
本发明中披露的数据是在小鼠的研究中获得的,但这些结论与人类同样相关,因为控制能量代谢的主要激素途径在小鼠和人类之间是类似的,因为它们显示出类似的受体表达谱。
[0049]
本发明的缀合物可以药物组合物的形式施用。因此,本发明进一步提供药物组合物,其包含本发明的缀合物或其药学上可接受的盐以及药学上可接受的载体。药物配制剂可通过常规技术制备。简言之,药学上可接受的载体可以是固体或液体。固体形式制剂包括粉末、片剂、丸剂、胶囊、扁囊剂、栓剂和分散颗粒。固体载体可以是一种或多种赋形剂,其也可以用作稀释剂、增溶剂、润滑剂、悬浮剂、粘合剂、防腐剂、润湿剂、片剂崩解剂或封装材料。
[0050]
包含在该药物配制剂中的缀合物可以处于粉末形式,通过无菌分离无菌固体或通过从在使用之前用合适的媒介物(例如无菌、无热原水)配制的溶液中冷冻干燥而获得。
[0051]
在一个实施方案中,该药物组合物适合于皮下施用、肌肉内施用、腹膜内施用、静脉内施用或口服施用。因此,本发明的组合物可以在安瓿、预填充注射器、小体积输注或多剂量容器中以单位剂量形式提供,任选地添加防腐剂。这些组合物可以采取在油状或水性媒介物中的悬浮液、溶液或乳液等形式。
[0052]
根据本披露内容,提供了药物组合物,其中将具有glp-1r活性的肽的降低食物摄入的作用与nmdar拮抗剂以单一方式结合。通过具有glp-1r活性的肽向下丘脑核、最后区、孤束核和腹侧被盖区和/或内分泌胰腺的主动递送将典型nmdar介导的不良神经生物学作用(比如解离症、精神病行为作用)与积极的代谢作用隔离开来。nmdar拮抗剂引起的不良神经生物学作用可包括幻觉、偏执妄想、混乱、注意力难以集中、激动、情绪改变、噩梦、紧张、共济失调、麻醉以及学习和记忆缺陷。nmdar拮抗剂的积极代谢作用可包括改善葡萄糖代谢、减少食物摄入和抑制暴食症,这可能有助于降低人类或哺乳动物的肥胖症和肥胖相关代谢障碍。
[0053]
因此,本发明的肽和nmdar拮抗剂配对的治疗效用为治疗肥胖症及其相关代谢障碍提供了新的途径。肥胖症的治疗可以由通过向人或哺乳动物施用缀合分子来降低食物摄入和食物动机以及减轻暴饮暴食发作来实现,并且因此,本发明的进一步的方面涉及一种降低哺乳动物体重的方法,该方法包括施用本发明的缀合分子或本发明的药物组合物。
[0054]
在一个实施方案中,减轻体重的方法需要通过向哺乳动物施用本发明的缀合分子或本发明的药物组合物来降低该哺乳动物的食物摄入。
[0055]
该缀合分子或该药物组合物可以皮下、口服、肌肉内、腹膜内或静脉内施用。
[0056]
与现有技术相比,该缀合分子以及该药物组合物在抑制食物摄入方面更为优越。因此,该缀合分子和该药物组合物可用于治疗任何程度的肥胖症。肥胖症可以按照体重指数(bmi)来描述,bmi定义为体重除以身高的平方,例如以kg/m2为单位来表示。在不受理论约束的情况下,诸位发明人认为bmi可用于定义致病性肥胖症和非致病性肥胖症之间的界限。例如,在本发明的上下文中,30kg/m2的bmi可以被解释为致病性肥胖症和非致病性肥胖症之间的界限。然而,也可以考虑bmi的其他值来定义致病性肥胖症和非致病性肥胖症之间
的界限。因此,例如,考虑了24kg/m2、26kg/m2、27kg/m2、28kg/m2、29kg/m2、30kg/m2、31kg/m2、32kg/m2、33kg/m2、34kg/m2和35kg/m2的bmi值来定义致病性肥胖症和非致病性肥胖症之间的界限。在进一步的方面,本发明涉及对哺乳动物进行的用于降低体重的非治疗性处理,其包括向所述哺乳动物口服施用根据本发明的缀合分子。例如,该哺乳动物可能具有非致病性bmi。特别地,该方法可包括向具有低于定义非致病性肥胖症的界限的bmi的受试者口服施用该缀合分子。
[0057]
在上文中,已经参考几个实施方案主要描述了本发明。然而,如本领域技术人员容易理解的,除上文披露的实施方案之外的其他实施方案在本发明的范围内同样是可能的。
[0058]
本发明的其他方面和有利特征在下面通过非限制性工作实施例详细描述和说明。
[0059]
通常,除非另有明确定义或说明,否则本文使用的所有术语将根据其在技术领域中的普通含义来解释,并且适用于本发明的所有方面和实施方案。除非另有明确说明,否则对所有提及
″
一个/一种/该[缀合物、分子、接头、肽等]
″
的内容均应公开解释为至少提及所述缀合物、试剂、分子、接头、肽等的至少一个实例。
[0060]
在本发明的上下文中,术语
″
glp1
″
、
″
glp-1
″
或
″
glp1肽
″
是指胰高血糖素超家族的肽,特别是肠促胰岛素激素胰高血糖素样肽1。本发明的肽也可以被认为是食物摄入调节激素肽,且作为本发明的缀合分子向下丘脑和/或胰腺的活性递送剂。
[0061]
在本发明的上下文中,术语
″
肽
″
是指由通过肽键连接的10至60个氨基酸构成的化合物。
[0062]
在本发明的上下文中,衍生自glp-1的肽是指与天然glp-1肽(即其所源于的seq id no:1)具有氨基酸序列同一性的肽。
[0063]
本文中所用的与肽或氨基酸相关的术语
″
衍生物
″
是指化学修饰的肽或氨基酸(其中在未修饰的肽、氨基酸或其类似物中不存在至少一个取代基),即已被共价修饰的肽和氨基酸。典型的修饰是酰胺、碳水化合物、烷基基团、酰基基团、酯等。
[0064]
在本发明的上下文中,术语
″
百分比同一性
″
或
″
%同一性
″
是指两个被比较的肽之间同一氨基酸的%,特别是使用blast算法。
[0065]
本文中使用的术语
″
nmdar拮抗剂
″
是指作为nmda受体(nmdar)的拮抗剂的化合物。nmdar拮抗剂的实例包括但不限于美金刚、盐酸美金刚、金刚烷胺、氯胺酮或mk801。nmdar拮抗剂的其他实例包括但不限于去甲氯胺酮和奈拉美生。
附图说明
[0066]
参考附图,通过以下对本发明实施方案的说明性而非限制性的详细描述,更好地理解本发明的上述以及附加目的、特征和优点,在附图中:
[0067]
图1显示了肽和nmdar拮抗剂缀合物的实例,
[0068]
图2显示了mk801从图1的缀合物释放的机制。
[0069]
图3显示了图1和图2的缀合物的三种形式的体外人血浆稳定性,
[0070]
图4显示了seq id no:1的肽和美金刚(glp-1 cys40/美金刚)的缀合物的减重作用,
[0071]
图5显示了glp-1 cys40/美金刚对小鼠累积食物摄入的作用,
[0072]
图6显示了glp-1 cys40/美金刚对小鼠每日食物摄入的作用,
[0073]
图7显示了glp-1 cys40/美金刚对小鼠身体成分的作用,
[0074]
图8显示了seq id no:1的肽和mk801(glp-1 cys40/mk801)的缀合物的减重作用,
[0075]
图9显示了glp-1 cys40/mk801缀合物对小鼠累积食物摄入的作用,
[0076]
图10显示了glp-1 cys40/mk801缀合物对小鼠每日食物摄入的作用,
[0077]
图11显示了glp-1 cys40/mk801缀合物对小鼠身体成分的作用,
[0078]
图12显示了seq id no:1的肽和mk801(glp-1 pen40/mk801)的缀合物的减重作用,其中seq id no:1中的半胱氨酸残基已被l-青霉胺取代,
[0079]
图13显示了glp-1 pen40/mk801缀合物对小鼠每日食物摄入的作用,且
[0080]
图14显示了glp-1 pen40/mk801缀合物对小鼠体重的作用。
[0081]
图15显示了化学接头衍生化的美金刚的合成路线。
[0082]
图16显示了肽和具有氨基基团的小分子缀合的示例合成路线。
[0083]
图17显示了合成化学接头衍生化的mk801的合成路线。
[0084]
图18显示了接头衍生化的mk801与肽(具有seq id no:1给出的氨基酸序列的肽)的缀合反应。
[0085]
图19显示了接头衍生化的mk801的化学合成路线。
[0086]
图20显示了接头衍生化的mk801与具有seq id no:1氨基酸序列且具有pen40修饰的肽的缀合反应。
[0087]
图21显示了不同剂量的glp-1 pen40/mk801缀合物对小鼠体重的作用。
[0088]
图22显示了不同剂量的glp-1 pen40/mk801缀合物对小鼠每日食物摄入的作用。
[0089]
图23显示了化合物耐受试验后不同剂量的glp-1 pen40/mk801缀合物对小鼠血糖的作用。
[0090]
图24显示了glp-1 pen40/mk801缀合物中有活性和无活性mk801对小鼠体重的作用。
[0091]
图25显示了glp-1 pen40/mk801缀合物中有活性和无活性mk801对小鼠累积食物摄入的作用。
[0092]
图26显示了用于缀合物glp-1 pen40/mk801的有活性和无活性mk801的体外人血浆稳定性。
[0093]
图27显示了具有不同接头的glp-1/mk801缀合物对小鼠体重的作用。
[0094]
图28显示了具有不同接头的glp-1/mk801缀合物对小鼠累积食物摄入的作用。
[0095]
图29是具有一种类型接头的glp-1/mk801缀合物。
[0096]
图30是具有一种类型接头的glp-1/mk801缀合物。
[0097]
图31显示了glp-1 pen40/mk801缀合物对小鼠蔗糖摄入的作用。
[0098]
图32显示了化合物耐受试验后glp-1 pen40/mk801缀合物对db/db小鼠血糖的作用。
[0099]
图33显示了协同激动剂gip/glp-1/mk801缀合物对小鼠体重的作用。
[0100]
图34显示了协同激动剂gip/glp-1/mk801缀合物对小鼠累积食物摄入的作用。
[0101]
图35显示了用于药物缀合物中的seq id no:9的协同激动剂glp-1/gip和seq id no:1的glp-1肽之间的氨基酸序列比对,其中x1是d-丙氨酸、d-丝氨酸、α-氨基异丁酸、n-甲基丝氨酸、甘氨酸或缬氨酸,且x2是半胱氨酸(hcys40/cys40)或l-青霉胺(pen40)。
[0102]
图36显示了与glp-1 pen40缀合的不同ndmar拮抗剂对小鼠体重的作用。
[0103]
图37显示了与glp-1 pen40缀合的不同ndmar拮抗剂对小鼠每日食物摄入的作用。
[0104]
图38显示了与glp-1 pen40缀合的不同ndmar拮抗剂对小鼠累积食物摄入的作用。
具体实施方式
[0105]
图1显示了肽和nmdar拮抗剂缀合物100的实例,其由mk801 101组成,mk801 101通过化学接头104化学附加到seq id no:1 103的肽的c端半胱氨酸102,该化学接头104包含二硫化物基团105。该c末端半胱氨酸102的侧链106可任选地衍生化,使得该侧链106的长度n为1或2个碳原子和/或r为氢或甲基。该侧链106的称为hcys40的修饰的长度n=2个碳原子且r=氢。该侧链106的称为hcys40的修饰的长度n=1个碳原子且r=甲基。常规半胱氨酸被称为cys40。
[0106]
图2显示了mk801从图1的缀合物100释放的机制。包含二硫化物基团105的化学接头104是自毁的,且可以在比如细胞内环境等还原环境(未示出)中还原以产生硫醇基团,从而将缀合物107的肽部分与缀合物的mk801部分108分离。在该分子的mk801部分108上,释放的亲核硫醇109经历自发的分子内环化,以释放mk801,作为天然未修饰的mk801药物(mk801的游离形式)。
[0107]
图3显示了图1和图2的缀合物100的三种形式的体外人血浆稳定性,每个形式具有不同的半胱氨酸衍生物或残基。第一形式glp-1 pen40/mk801具有半胱氨酸衍生物pen40,第二形式glp-1 hcys40/mk801具有胱氨酸衍生物hcys40,第三形式glp-1 cys40/mk801具有未修饰的半胱氨酸cys40。每个形式的血浆稳定性显示为随时间的回收率百分比。lcms分析(未示出)揭示,对缀合物降解的主要贡献来源于mk801的脱缀合,可能是通过该接头的二硫化物交换。因此,该c末端半胱氨酸102(hcys40/cys40)对l-青霉胺(pen40)的单取代由于空间位阻增加而降低了二硫键的可及性,从而显著提高了血浆稳定性。
[0108]
图4-14显示了实施例8中披露的体内小鼠研究的结果。
[0109]
图4显示了通过图1和2中所示的接头化学附加的seq id no:1的肽与美金刚的缀合物(其中该半胱氨酸残基是未修饰的半胱氨酸)(glp-1-cys40/美金刚)(40nmol/kg)和等摩尔剂量的seq id no:1的肽(glp-1cys40)或美金刚(该等摩尔剂量以治疗8天的饮食诱导(dio)小鼠的体重百分比(bw%)测量)的减重作用。数据表示为平均值
±
sem,并且每组n为8。glp-1 cys40和glp-1 cys40/美金刚均导致dio小鼠的bw%降低,后一种缀合物导致治疗8天后大约7%的bw%降低。
[0110]
图5显示了glp-1 cys40/美金刚和等摩尔剂量的glp-1 cys40或美金刚对治疗8天的dio小鼠的累积食物摄入(累积fi,克/天)的作用。数据表示为平均值
±
sem,并且每组n=8。在治疗过程中,与对照(媒介物)和美金刚相比,在用glp-1 cys40和glp-1 cys40/美金刚治疗的小鼠中观察到累积食物摄入降低。
[0111]
图6显示了glp-1 cys40/美金刚(40nmol/kg)或等摩尔剂量的glp-1 cys40或美金刚对治疗8天的dio小鼠的每日食物摄入(每日fi,克/天)的作用。数据表示为平均值
±
sem,并且每组n=8。在8天的治疗期间,与美金刚治疗的小鼠和对照组(媒介物,即盐水)相比,glp-1 cys40和glp cys40/美金刚通常显示每日食物摄入降低。研究结束时,与对照组(媒介物)相比,用glp-1治疗的小鼠的食物摄入仅略有降低。
[0112]
图7显示了glp-1 cys40/美金刚(40nmol/kg)或等摩尔剂量的glp-1 cys40或美金刚对治疗8天的dio小鼠的身体成分(δ变化,g)(在脂肪量和瘦体重变化方面)的作用。数据表示为平均值
±
sem,并且每组n=8。8天后,用美金刚、glp-1 cys40和glp-1 cys40/美金刚治疗的小鼠均显示体脂量降低,而瘦体重几乎没有变化。glp-1 cys40/美金刚导致最高的体脂量变化,在用该缀合物治疗的小鼠中观察到大约4g脂肪量降低。
[0113]
图8显示了在治疗10天的dio小鼠中,glp-1 cys40/mk801(100nmol/kg)或等摩尔剂量的glp-1 cys40或mk801的减重作用(bw%)。数据表示为平均值
±
sem,并且每组n=8。在治疗10天后,mk801几乎没有显示体重(bw)百分比变化,而glp-1 cys40和glp-1 cys40/mk801分别导致bw降低大约8%和12%。
[0114]
图9显示了glp-1 cys40/mk801(100nmol/kg)或等摩尔剂量的glp-1 cys40或mk801对治疗10天的dio小鼠的累积食物摄入(累积fi)的作用。数据表示为平均值
±
sem,每组n=8。在10天的治疗过程中,与对照(媒介物)和mk801相比,在用glp-1 cys40和glp-1 cys40/mk801治疗的小鼠中观察到累积食物摄入降低。对glp-1 cys40/mk801治疗的小鼠观察到最佳结果,其累积食物摄入大约为13g/天,这比媒介物治疗的小鼠(大约23g/天)少大约10g/天。
[0115]
图10显示了glp-1 cys40/mk801(100nmol/kg)或等摩尔剂量的glp-1 cys40或mk801对治疗10天的dio小鼠的每日食物摄入(每日fi)的作用。数据表示为平均值
±
sem,每组n=8。通常来说,在10天的治疗期间,每日食物摄入在不同程度上波动,然而,与对照组(媒介物)相比,用glp-1 cys40/mk801治疗的小鼠在10天内都观察到食物摄入降低。
[0116]
图11显示了glp-1 cys40/mk801(100nmol/kg)或等摩尔剂量的glp-1 cys40或mk801对治疗10天的dio小鼠身体成分(δ变化,g)(在脂肪量和瘦体重变化方面)的作用。数据表示为平均值
±
sem,并且每组n=8。在10天的治疗后,glp-1 cys40/mk801治疗的小鼠组显示出脂肪量和瘦体重的降低,其中脂肪量的变化(降低几乎5g)最为显著。
[0117]
图12显示了glp-1 pen40/mk801(100nmol/kg)或等摩尔剂量的glp-1 cys40或mk801对治疗5天的dio小鼠的体重%的作用。数据表示为平均值
±
sem,每组n=8。治疗5天后,glp-1 pen40/mk801治疗的小鼠显示出大约15%的体重降低。相比之下,glp-1 cys40治疗的小鼠显示出大约4%的体重降低。
[0118]
图13显示了glp-1 pen40/mk801(100nmol/kg)或等摩尔剂量的glp-1 cys40或mk801对治疗5天的dio小鼠的食物摄入(g/天)的作用。数据表示为平均值
±
sem,每组n=8。与对照组(媒介物治疗的小鼠)相比,用glp-1 pen40/mk801治疗的小鼠显示出食物摄入的快速降低。此外,在5天的治疗期间,降低的食物摄入维持在0.2g/天-0.7g/天左右。
[0119]
图14显示了glp-1 pen40/mk801(100nmol/kg)或等摩尔剂量的glp-1 pen40或glp-1 cys40对治疗5天的dio小鼠的体重%的作用。数据表示为平均值
±
sem,每组n=7。用glp-1 pen40或glp-1 cys40治疗的小鼠显示出类似的体重%降低(大约6%),而glp-1 pen40/mk801显示出大约12%的体重降低。此外,根据曲线斜率,我们会看到如果延长治疗,glp-1 pen40/mk801的情况可以预期到体重的进一步降低。
[0120]
图21和22显示了与对照组(媒介物,即盐水)相比,不同剂量(50nmol/kg和100nmol/kg)的glp-1 pen40/mk801缀合物对治疗5天的dio小鼠的体重(bw%,图21)和每日食物摄入(每日fi,单位:克,图22)的作用。数据表示为平均值
±
sem,每组n=5-6。在治疗过
程中,与对照组相比,观察到两种剂量(50nmol/kg和100nmol/kg)治疗的小鼠体重和每日食物摄入降低,每日皮下注射100nmol/kg缀合物的小鼠体重降低最为显著。
[0121]
图23显示了与对照组(媒介物,即盐水)相比,不同剂量(50nmol/kg和100nmol/kg)的glp-1 pen40/mk801缀合物对治疗过程第7天接受ipgtt的dio小鼠的血糖水平(mmol/l)的作用。在120分钟的过程中测量血糖水平。数据表示为平均值
±
sem,每组n=5-6。通常,与对照组相比,两种剂量(即50nmol/kg和100nmol/kg)的缀合物导致显著更低的初始增加和总体更低的血糖水平。
[0122]
图24和25显示了与对照组(媒介物,即盐水)相比,与glp-1 pen40缀合的有活性和无活性mk801对治疗7天的dio小鼠的体重(δ体重%,图24)和累积食物摄入(累积fi,克,图25)的作用。数据表示为平均值
±
sem,每组n=8。在治疗过程中,观察到用与有活性mk801缀合的glp-1 pen40治疗的小鼠体重和累积食物摄入降低。与无活性mk801的缀合物显示出与未缀合glp-1 pen40类似的结果。结论是mk801和glp-1在降低小鼠体重和累积食物摄入方面具有协同作用。
[0123]
图26显示了与pbs对照相比,缀合物glp-1 pen40/mk801的有活性和无活性mk801形式的体外人血浆稳定性。无活性和有活性mk801的血浆稳定性显示为随时间(小时)的回收率百分比(%)。与mk801是有活性的还是无活性的无关,这两种缀合物显示出几乎相同的血浆稳定性。
[0124]
图27和28显示了在治疗7天的dio小鼠中,与对照组(媒介物,即盐水)相比,具有不同接头的glp-1/mk801缀合物(100nmol/kg)对体重(bw%,图27)和累积食物摄入(累积fi,克,图28)的作用。数据表示为平均值
±
sem,每组n=5-6。图20(glp-1 pen40/mk801)、图29(glp-1 lys40-三唑-peg
4-val-cit-pab-mk801)和图30(glp-1 cys40-mc-val-cit-pab-mk801)显示了具有不同接头的glp-1/mk801缀合物的结构。在治疗过程中,用具有不同接头的glp-1/mk801缀合物治疗的小鼠显示出累积食物摄入降低。在用glp-1 pen40/mk801缀合物治疗的小鼠组中,观察到在治疗的7天内体重降低最为显著(大约20%降低)。
[0125]
图31显示了与对照组(媒介物,盐水注射)相比,glp-1 pen40/mk801(100nmol/kg)和等摩尔剂量的glp-1 pen40、mk801或索马鲁肽对治疗8天的dio小鼠的蔗糖摄入(以%计)的作用。数据表示为平均值
±
sem,每组n=8。与对照组(媒介物)相比,观察到用索马鲁肽和glp-1/mk801缀合物治疗的小鼠的蔗糖摄入降低最为显著。得出的结论是,本发明的缀合分子在所治疗小鼠中有效地诱导食物奖赏和饱腹感。
[0126]
图32显示了glp-1 pen40/mk801缀合物(100nmol/kg)和等摩尔剂量的mk801或索马鲁肽对治疗过程第7天接受ipgtt的db/db(糖尿病)小鼠的血糖(mmol/l)的作用。在24小时的过程中测量血糖水平。数据表示为平均值
±
sem,每组n=8。与对照组(媒介物)相比,用索马鲁肽或缀合物glp-1 pen40/mk801治疗的小鼠显示出总体较低的血糖水平,并且得出的结论是,本发明的缀合分子适合于治疗糖尿病小鼠。
[0127]
图33和34显示了与对照组(媒介物,盐水注射)相比,协同激动剂gip/glp-1 pen40/mk801缀合物(seq id no:9)(50nmol/kg)和等摩尔剂量的glp-1/gip对治疗7天的dio小鼠的体重(以%计,图33)和累积食物摄入(以克计的累积fi,图34)的作用。数据表示为平均值
±
sem,每组n=8。在用gip/glp-1/mk801缀合物治疗的小鼠中观察到最显著的作用,与对照组相比,这些小鼠显示体重总体降低大约25%,与对照组观察到的15g累积食物
摄入相比,这些小鼠显示大约3g累积食物摄入。
[0128]
图36至38显示了与对照组(媒介物,即盐水)相比,与glp-1 pen40(100nmol/kg)缀合的不同ndmar拮抗剂(即mk-801、美金刚和奈拉美生)对治疗5天的dio小鼠的体重(%,图36)、每日食物摄入(食物摄入,克/天,图37)和累积食物摄入(累积fi,克,表38)的作用。数据表示为平均值
±
sem,每组n=8。在治疗过程中,与对照组相比,用与mk801、美金刚或奈拉美生缀合的glp-1 pen40治疗的小鼠均显示体重显著降低以及每日和累积食物摄入降低。得出的结论是,可以将不同的nmdar拮抗剂与本发明的肽缀合,以获得对小鼠体重和食物摄入的相同有益效果。
[0129]
结论
[0130]
所提供的数据表明,glp-1类似物和nmdar拮抗剂的化学缀合代表了有效逆转肥胖症的新的药物策略。与glp-1肽对照相比,基于该策略的缀合物在抑制食物摄入和减轻体重方面具有优势,并且在nmdar拮抗的不利中枢作用方面没有缺陷。
[0131]
实施例
[0132]
实施例1:肽和肽-nmdar拮抗剂缀合物的制备。
[0133]
材料:所有溶剂和试剂均购自商业来源,使用时无需进一步纯化。将h-rink酰胺树脂用于肽延伸。除非另有说明,fmoc保护的(9-芴基甲基氨基甲酸酯)氨基酸购自爱丽思生物技术公司(iris biotech)或陀螺仪蛋白质技术公司(gyros protein technologies),并且h-rink酰胺树脂(35-100目;上样0.40mmol/g-0.60mmol/g)购自西格玛奥德里奇公司(sigma-aldrich)。市售的nα-fmoc氨基酸结构单元作为以下侧链保护的类似物购买:arg,pmc;asp,o
t
bu;cys,trt;gln,trt;his,trt;lys,trt;ser,
t
bu;和trp,boc(pmc=2,2,5,7,8-五甲基色满-6-磺酰基,o
t
bu=叔丁酯,trt=三苯甲基,boc=叔丁氧基羰基,并且
t
bu=叔丁醚)。
[0134]
所有肽和肽与nmdar拮抗剂的缀合物均通过以下进行表征:分析型反相超高效液相色谱(rp-uplc)(沃特斯公司(waters))和电喷雾电离液相色谱质谱(esi-lcms)偶联安捷伦公司(agilent)6410三重四极滤质器(具有c18柱,zorbax eclipse,xbd-c18,4.6
×
50mm)。将esi-lcms用由h2o∶mecn∶tfa(a:95∶5∶0.1,b:5∶95∶0.1)构成的二元缓冲液体系以0.75ml/min的流速洗脱。通过配备c18柱(acquity uplc beh c18,1.7μm,2.1
×
50mm)的rp-uplc测定纯度,用h2o∶mecn∶tfa(a:95∶5∶0.1,b:5∶95∶0.1)构成的二元缓冲液体系以0.45ml/min的流速洗脱。
[0135]
fmoc保护方案的自动肽合成方案:使用prelude x感应加热辅助肽合成仪(陀螺仪蛋白质技术公司,图森,亚利桑那州,美国)用10ml玻璃容器作为其c末端酰胺化衍生物来制备肽。所有试剂作为在dmf中的原液新鲜制备:fmoc保护的氨基酸(0.2m)、hctu(0.5m)、dipea(1.0m)和哌啶(20%v/v)。使用以下方案通过连续的合成操作实现肽延伸:脱保护(2
×
2min,rt,300rpm摇动)和偶联(2
×
5min,75℃,300rpm摇动,对于arg和his 2
×
5min,50℃,300rpm摇动)。使用由与树脂相比过量5倍的aa/hctu/dipea(比率1∶1.25∶2.5)组成的双偶联和三偶联制备肽。
[0136]
肽裂解:通过每100mg肽基树脂加入1.5ml裂解混合物(在tfa中2.5%edt、2.5%h2o、2.5%tips、2.5%茴香硫醚),随后搅拌2小时,从肽基树脂中释放合成的肽。将粗肽在冷乙醚中沉淀,在4℃下以2500
×
g离心10min,重新溶解在mecn∶h2o∶tfa(比率1∶1∶0.01),
过滤并冷冻干燥。
[0137]
纯化:在纯化之前,通过rp-uplc和esi-lcms或maldi-tof质谱法分析粗肽或肽与nmdar拮抗剂的缀合物。使用配备有反相c18柱(zorbax,300sb-c18,21.2
×
250mm)的反相高效液相色谱(rp-hplc)系统(沃特斯公司)进行纯化,并使用h2o∶mecn∶tfa(a:95∶5∶0.1;b:5∶95∶0.1)的二元缓冲液系统以线性梯度(流速20ml/min)洗脱。以0.3分钟的间隔收集级分并进行esi-lcms表征。通过rp-uplc在214nm处测定纯度,将纯度>95%的级分合并并冷冻干燥。将最终的冷冻干燥产物用于进一步的实验。
[0138]
肽和nmdar拮抗剂的缀合物组装的缀合方案:将纯肽和纯硫代吡啶基激活的nmdar拮抗剂缀合物溶解在二元溶剂系统(a:dmf;在h2o中6m胍、1.5m咪唑,ph=8)(比率7∶1)中并搅拌至少2小时。通过分析型rp-uplc和esi-lcms监测粗反应混合物。完成后,用缓冲液a和缓冲液b稀释反应混合物,并使用rp-hplc以线性梯度洗脱直接纯化。
[0139]
脱盐:在生物实验之前,所有肽都被脱盐。通过将肽或肽与nmdar拮抗剂的缀合物连续重新溶解在0.01m稀hcl水溶液中,随后冷冻干燥,重复3次来进行脱盐。在用于体内或体外实验之前,通过rp-uplc和esi-lcms监测该肽或该缀合物的纯度。
[0140]
glp-1cys40/美金刚(半胱氨酸连接)的制备。
[0141]
使用如上所述的fmoc方案合成氨基酸序列为seq id no:1的glp-1肽,并将其与化学接头衍生化的美金刚类似物缀合。通过图15所示的合成路线合成化学接头衍生化的美金刚。合成路线的第一步在室温下在meoh中进行2小时。第二步在0℃下在吡啶存在下在ch2cl2中进行2小时。第三步在55℃下在n,n-二异丙基乙胺(dipea)存在下在dmf中进行5天。最后一步(缀合)在室温下在6m胍、1.5m咪唑缓冲液中进行2小时。
[0142]2′‑
吡啶基二硫代乙醇。在配备有磁力搅拌棒的干燥圆底烧瓶中,在n2气氛下,将2
′‑
aldrithiol(4.71g,21.3mmol,3当量)溶解在无水meoh(20ml)中,随后通过注射器滴加2-巯基乙醇(0.56g,7.1mmol,0.5ml,1当量)。将反应在环境温度下放置2小时,然后真空浓缩。通过硅胶快速色谱(etoac:ch2cl2,2:8)纯化粗黄色油,得到呈透明油的2
′‑
吡啶基二硫代乙醇(1.33g,100%)。rf=0.48;1h nmr(600mhz,氯仿-d)δ8.49(d,j=5.0hz,1h),7.57(td,j=7.7,1.8hz,1h),7.44-7.36(m,1h),7.16-7.11(m,1h),5.32(s,1h),3.88-3.73(m,2h),3.01-2.89(m,2h);
13
c nmr(151mhz,cdcl3)δ159.31,149.86,137.00,122.12,121.57,58.37,42.83。
[0143]
4-硝基苯基(2-(吡啶-2-基二硫基)乙基)碳酸酯。在配备有磁力搅拌棒的干燥圆底烧瓶中,并且在n2气氛下,将2
′‑
吡啶基二硫代乙醇(1.33g,7.1mmol,1当量)和无水吡啶(0.56g,8.5mmol,0.575ml,1.2当量)在无水ch2cl2(15ml)中稀释。将反应混合物冷却至0℃,一次性加入氯甲酸硝基苯基酯(1.72g,8.5mmol,1.2当量)。将反应搅拌10分钟,使其达到环境温度,并在搅拌下放置2小时。将反应稀释至50ml,并用3x h2o(30ml)和盐水(30ml)萃取,用mgso4干燥,过滤并真空浓缩。通过硅胶快速色谱(庚烷:etoac,2:1)纯化呈油,得到呈透明粘性油的4-硝基苯基(2-(吡啶-2-基二硫基)乙基)碳酸酯(2.21g,89%)。rf=0.34;纯度>95%(hplc),r
t
=15.99min;uplc/ms(esi):对于c
14h12
n2o5s2[m h]
=353.0的m/z计算值,发现353.3m/z;1h nmr(600mhz,dmso-d6)δ8.47(ddd,j=4.8,1.9,0.9hz,1h),8-35-8.26(m,2h),7.84(td,j=7.8,1.8hz,1h),7.78(dt,j=8.1,1.1hz,1h),7.58-7.48(m,2h),7.26(ddd,j=7.3,4.8,1.1hz,1h),4.48(t,j=6.0hz,2h),3.24(t,j=6.1hz,2h);
13
c nmr
(151mhz,dmso)δ158.65,155.17,151.75,149.66,145.18,137.80,125.40,122.53,121.40,119.52,66.54,36.42。
[0144]
2-(吡啶-2-基二硫基)乙基(3,5-二甲基金刚烷-1-基)氨基甲酸酯在配备有磁力搅拌棒的干燥圆底烧瓶中,并且在n2下,将4-硝基苯基(2-(吡啶-2-基二硫基)乙基)碳酸酯(707mg,2.00mmol,1当量)和盐酸美金刚(650mg,3.00mmol,1.5当量)溶解在无水dmf(20ml)中,并通过注射器加入无水dipea(260mg,6.00mmol,0.35ml,3当量)。美金刚未完全溶解,并且在加入dipea后,反应立即变黄。将反应放置5天,随后加热至80℃。然后将反应用etoac(50ml)转移到分液漏斗中,并用5x半饱和盐水(50ml)和盐水(50ml)彻底洗涤以除去dmf。随后用5x1m naoh水溶液(50ml)萃取有机层(直到水层的黄色停止),用mgso4干燥,过滤并真空浓缩。通过硅胶快速色谱纯化粗油,以梯度(庚烷:etoac,9:1至3:1)洗脱,得到呈玻璃状粘性油的2-(吡啶-2-基二硫基)乙基(3,5-二甲基金刚烷-1-基)氨基甲酸酯(540mg,54%)。rf=0.26;纯度>95%(hplc),r
t
=19.36min;uplc/ms(esi):对于c
20h28
n2o2s2[m h]
=393.2的m/z计算值,发现393.4m/z;1h nmr(600mhz,dmso-d6)δ8.46(ddd,j=4.8,1.9,0.9hz,1h),7.85-7.75(m,2h),7.25(ddd,j=7.2,4.8,1.2hz,1h),6.89(s,1h),4.10(t,j=6.4hz,2h),3.05(t,j=6.3hz,2h),1.69-1.63(m,2h),1.54-1.43(m,4h),1.31-1.20(m,5h),1.07(s,2h),0.80(s,6h);
13
c nmr(151mhz,dmso)δ159.04,153.78,149.55,137.79,121.21,119.23,60.80,51.40,50.18,47.07,42.22,37.46,31.84,30.05,29.46。
[0145]
使用上述方案制备glp-1 cys40和glp-1 cys40/美金刚。rp-uplc和esi-lcms分析确定纯度>95%。
[0146]
glp-1 pen40/美金刚(青霉胺连接)的制备。
[0147]
使用图15中披露的合成路线进行化学接头衍生化的美金刚的合成。glp-1 pen40和美金刚通过图16所示的化学反应缀合,该化学反应在室温下在6m胍、1.5m咪唑缓冲液中进行2小时。
[0148]
glp-1 cys40/mk801(半胱氨酸连接)的制备。
[0149]
使用上文披露的fmoc方案合成序列为seq id no:1的肽,并将其与化学接头衍生化的mk801类似物缀合。通过图17中披露的第二合成路线进行化学接头衍生化的mk801的合成。化学反应在55℃下在dipea存在下在dmf中进行5天。
[0150]
通过图18所示的化学反应将接头衍生化的mk801与glp-1 cys40缀合。反应在室温下在6m胍、1.5m咪唑缓冲液中进行2小时。
[0151]
2-(吡啶-3-基二硫基)乙基5-甲基-10,11-二氢-5h-5,10-桥亚胺二苯并[a,d][7]轮烯-12-甲酸酯。在配备有磁力搅拌棒的火焰干燥的schlenk圆底烧瓶中,在n2气氛下,将mk801盐酸盐191mg,0.86mmol,1.2当量)溶解在无水dmf(10ml)中,随后加入4-硝基苯基(2-(吡啶-2-基二硫基)乙基)碳酸酯(253mg,0.72mmol,1.0当量)。随后,加入无水dipea(375μl,2.14mmol,3.0当量),溶液变黄。将反应在油浴中加热至55℃并搅拌4天,直到uplc-ms指示原料完全消耗。用etoac(50ml)稀释反应,并用半饱和盐水(5x60ml)、0.5mnaoh水溶液(5x60ml)和盐水彻底洗涤。收集有机层,用mgso4干燥,过滤并真空浓缩。通过制备型hplc纯化(以等度60%b洗脱,超过17ml/min),随后冷冻干燥,得到呈透明固体的11(250.2mg,80.1%);纯度>95%(hplc),r
t
=18.17min;uplc/ms(esi):c
24h22
n2o2s2[m h]
=435.1的m/z计算值,发现435.4;1h nmr(600mhz,dmso-d6)δ8.41(dt,j=4.8,1.4hz,1h),7.68(dt,j=
7.9,4.1hz,2h),7.45(d,j=7.1hz,1h),7.38-7.31(m,1h),7.25-7.15(m,4h),7.15-7.06(m,2h),7.01-6.87(m,1h),5.38(d,j=5.5hz,1h),4.27-4.13(m,2h),3.59(dd,j=17.3,5.7hz,1h),3.10(s,2h),2.67-2.58(m,1h),2.20(s,3h);
13
c nmr(151mhz,dmso)δ158.92,149.56,143.37,139.04,137.70,131.78,130.25,127.42,127.34,127.31,125.88,122.12,121.66,121.20,119.19,65.33,62.21,59.20,37.55。
[0152]
使用上文披露的方案由2-(吡啶-3-基二硫基)乙基5-甲基-10,11-二氢-5h-5,10-桥亚胺二苯并[a,d][7]轮烯-12-甲酸酯和glp-1 cys40制备glp-1 cys40/mk801。rp-uplc和esi-lcms分析证实该产物,并确定纯度>95%。
[0153]
glp-1 hcys40/mk801(同型半胱氨酸连接)的制备。
[0154]
使用上文披露的fmoc方案合成具有seq id no:1的氨基酸序列和hcys40修饰的肽,并将其与化学接头衍生化的mk801类似物缀合。通过图19所示的合成路线进行接头衍生化的mk801的化学合成,化学反应在室温下在6m胍、1.5m咪唑缓冲液中进行2小时。
[0155]
glp-1 hcys40:使用上文披露的方案制备具有seq id no:1的氨基酸序列和hcys40修饰的肽。rp-uplc和esi-lcms分析确定纯度>95%。使用上文披露的方案制备glp-1 hcys40/mk801。rp-uplc和esi-lcms分析确定纯度>95%。
[0156]
glp-1 pen40/mk801(青霉胺连接)的制备。
[0157]
使用上文披露的fmoc方案合成glp-1肽衍生物,并将其与化学接头衍生化的mk801类似物缀合。通过图16中披露的路线进行化学接头衍生化的mk801的化学合成。
[0158]
glp-1 pen40/mk801:使用上文披露的方案和图20所示的化学反应制备缀合物,该化学反应在室温下在6m胍、1.5m咪唑缓冲液中进行2小时。rp-uplc和esi-lcms分析确定纯度>95%。
[0159]
实施例2:体外人血浆稳定性研究。
[0160]
体外人血浆稳定性测定:使用含有柠檬酸磷酸葡萄糖的正常人血浆(3h生物医学公司(3h biomedical),批号p22)测定肽稳定性。将人血浆在37℃下预热15分钟。随后,将360μl人血浆中掺入40μl glp-1 pen40/mk801、glp-1 hcys40/mk801或glp-1 cys40/mk801缀合物原液(1mm,通过用pbs缓冲液由dmso原液中的10mm肽进行稀释而制备),并在37℃下在轻轻摇动下进行温育。在t=0和5个额外时间点(取决于缀合物的稳定性)收集45μl的等分试样,并在0℃下用脲缓冲液(50μl,30分钟)预处理,随后用丙酮中的20%三氯乙酸处理,并在-20℃下温育过夜。离心(13400rpm,30min)后,将上清液过滤并通过rp-uplc在214nm和esi-lcms进行分析。测定曲线下面积(auc)并使用prism 8.0绘图。通过将数据拟合到单相衰减方程来确定半衰期(t
1/2
)。数据表示为三次单独实验的平均值。
[0161]
实施例3:饮食诱导肥胖症(dio)小鼠体内药理学研究。
[0162]
c57bl6j雄性小鼠(以下称为饮食诱导肥胖症(dio)小鼠)维持高脂肪饮食(58%的能量来自脂肪),并且对于每项研究而言,研究开始前的平均体重超过45克。将小鼠单独圈养,抑或双只圈养。将小鼠维持在21℃-23℃的12h暗-光循环下。每日皮下施用一次化合物(下午2点至5点),并在相应时间测量食物摄入(fi)和体重(bw)。对于身体成分,在研究前(研究开始前1-3天)和研究的最后一天使用mri扫描仪(echomri)测量脂肪和瘦肉量。注射媒介物(盐水)的小鼠组作为对照组。
[0163]
实施例4:在食物喂养小鼠中的蔗糖偏好试验。
[0164]
c57bl6j雄性小鼠被单独圈养在笼子中,并维持食物饮食。除以10nmol/kg的剂量施用的索马鲁肽外,所有化合物以100nmol/kg的剂量每日皮下施用一次。注射媒介物(盐水)的小鼠组作为对照组。每个治疗组包括8只小鼠。所有笼子都配备有两个饮用瓶,小鼠在研究开始前至少适应五天。研究开始后,用一个含有水的瓶和一个含有10%(w/v)蔗糖水溶液的瓶替换水瓶。蔗糖瓶按左右瓶均等分布,以纠正侧偏。24小时后通过称量这些瓶来测量蔗糖水摄入和水摄入。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。