1.本发明涉及聚芳醚酮技术领域,尤其涉及一种膦酰亚胺缩聚单体、其制备方法、膦酰亚胺型前驱体聚合物、其制备方法与聚芳醚酮的制备方法。
背景技术:
2.聚芳醚酮(paeks)是一类高性能聚合物,普遍具有较高的耐热性能、良好的加工性、优异的机械性能、电绝缘性、耐化学性和尺寸稳定性,材料合成简单,性价比高,在航空和航天、汽车、电子、精密机械等领域被广泛关注。聚芳醚酮(paeks)类代表产品包括聚醚酮(pek)、聚醚醚酮(peek)以及聚醚酮酮(pekk)等。由于优良的性能,聚芳醚酮类聚合物广泛用于电子、电气、机械、医疗、汽车及航空航天等工业领域,是公认的高性能工程塑料。peek、pek等结构中,分子链中只有二苯甲酮与苯醚类的结构单元,分子链刚性且存在极强的π-π堆砌,所以该类聚芳醚酮具备非常优异的耐溶剂性、抗腐蚀性、抗氧化性等抗逆性能,但也代表了其只能通过熔融加工,难以通过溶剂方法加工成膜。
3.张所波等人(cn 110052178 a)利用含有苯亚胺席夫碱侧基的peek聚合物前驱体进行peek的溶液加工,即配置成铸膜液,相转化法制成不对称膜后,再将不对称膜置于酸液中酸化,膜中主链结构中的亚胺水解转化为酮,主链结构转化为性能优异的peek。如该专利当中的案例,peek本身无法利用溶剂加工,更无法配置成铸膜液,但引入的苯亚胺侧基阻碍了分子链间的堆砌使聚合物溶解度大增,加工成不对称膜后还可以通过化学反应除去侧基使分子链变为性能优异的peek,赋予制备的不对称膜优异的耐溶剂性。但是由苯胺与二卤代二苯酮合成的苯亚胺型结构虽然可以参与缩聚,但制备的聚合物前驱体在酸性条件下的水解速度较慢,而且该类聚合物的溶解度虽然得到了改善,但是针对不同的加工方式与应用场景,单一的结构仍然欠缺普适性与灵活性。
技术实现要素:
4.本发明解决的技术问题在于提供一种膦酰亚胺型前驱体聚合物,该前驱体聚合物可在酸条件下快速水解膦酰亚胺侧基以得到性能优异的聚芳醚酮。
5.有鉴于此,本技术提供了一种如式(ⅰ)所示的膦酰亚胺缩聚单体,
[0006][0007]
其中,x选自卤素;
[0008]
r1和r2独立的选自氰基、取代或未取代的c1~c10的烷基,取代或未取代的c1~c5的烯基、取代或未取代的c6~c20的芳基。
[0009]
优选的,所述r1和r2独立的选自以下并不限于以下基团中的一种:
[0010][0011]
本技术还提供了所述的如式(ⅰ)所示的的膦酰亚胺缩聚单体的制备方法,包括以下步骤:
[0012]
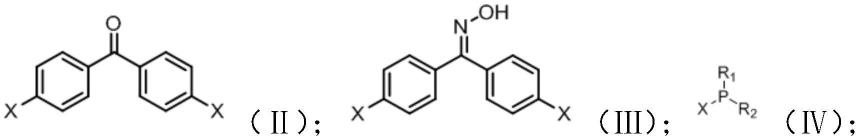
a)将如式(ii)所示的4,4
’‑
二卤二苯酮、盐酸羟胺和醋酸钠在有机溶剂中混合,加热回流,得到如式(ⅲ)所示的4,4
’‑
二卤二苯酮肟;
[0013]
b)将所述4,4
’‑
二卤二苯酮肟和如式(ⅳ)所示的卤化膦化合物在弱碱环境中反应,得到如式(ⅰ)所示的膦酰亚胺缩聚单体;
[0014][0015]
其中,x选自卤素;
[0016]
r1和r2独立的选自氰基、取代或未取代的c1~c10的烷基,取代或未取代的c1~c5的烯基、取代或未取代的c6~c20的芳基。
[0017]
优选的,步骤a)中,所述4,4
’‑
二卤二苯酮和盐酸羟胺的摩尔比为1:1.1~1:1.6,所述4,4
’‑
二卤二苯酮与所述醋酸钠的摩尔比为1:1.6~1:2.0;所述加热回流的时间为12~36h;所述有机溶剂为体积比为(6~1):1的乙醇和水的混合溶剂。
[0018]
优选的,步骤b)中,所述4,4
’‑
二卤二苯酮肟和所述弱碱的摩尔比为1:(1.1~1.5),所述反应的温度为-80~0℃,所述弱碱选自三乙胺、三甲胺和n,n-二异丙基乙胺中的一种或多种。
[0019]
本技术还提供了一种如式(
ⅴ
)的膦酰亚胺型前驱体聚合物,
[0020][0021]
其中,r1和r2独立的选自氰基、取代或未取代的c1~c10的烷基,取代或未取代的c1~c5的烯基、取代或未取代的c6~c20的芳基;
[0022]
r3选自取代或未取代的c5~c30的芳基或杂环芳基;
[0023]
n为聚合度。
[0024]
优选的,所述r3选自以下基团中的一种:
[0025][0026][0027]
本技术还提供了所述的如式(
ⅴ
)所示的膦酰亚胺型前驱体聚合物的制备方法,包括以下步骤:
[0028]
将如式(ⅵ)所示的二酚单体的醇溶液与氢氧化钠混合,反应,得到酚钠盐;
[0029]
将酚钠盐和如式(ⅰ)所示的膦酰亚胺缩聚单体反应,得到如式(
ⅴ
)所示的膦酰亚胺型前驱体聚合物;
[0030]
ho-r
3-oh(ⅵ);
[0031][0032]
其中,x选自卤素;
[0033]
r1和r2独立的选自氰基、取代或未取代的c1~c10的烷基,取代或未取代的c1~c5的烯基、取代或未取代的c6~c20的芳基;
[0034]
r3选自取代或未取代的c5~c30的芳基或杂环芳基;
[0035]
n为聚合度。
[0036]
优选的,所述膦酰亚胺型前驱体聚合物的制备方法具体为:
[0037]
a)将二酚单体的醇溶液与强极性非质子溶剂混合,在氮气保护下加入氢氧化钠固体,反应,得到酚钠盐;
[0038]
b)将步骤a)得到的反应体系加热至80~100℃以去除醇,再加入甲苯,加热至140~160℃,共沸除水;然后加入膦酰亚胺缩聚单体,加热至150~200℃,反应后得到膦酰亚胺型前驱体聚合物。
[0039]
本技术还提供了一种如式(ⅶ)所示的聚芳醚酮的制备方法,包括:
[0040]
将如式(
ⅴ
)所示的膦酰亚胺型前驱体聚合物与酸性溶液混合,水解,得到如式(ⅶ)所示的聚芳醚酮;
[0041][0042]
其中,r1和r2独立的选自氰基、取代或未取代的c1~c10的烷基,取代或未取代的c1~c5的烯基、取代或未取代的c6~c20的芳基;
[0043]
r3选自取代或未取代的c5~c30的芳基或杂环芳基;
[0044]
n为聚合度。
[0045]
本技术提供了一种如式(
ⅴ
)所示的膦酰亚胺型前驱体聚合物,其溶解度得到提高是由于主链上带有的大位阻侧基阻碍了分子链堆砌,因而利于聚芳醚酮类聚合物溶液加工;同时由于膦酰胺结构的高极性使得前驱体聚合物的侧基在酸性条件下可快速水解脱落,由此可制备得到性能优秀的聚芳醚酮类聚合物(paeks)。
附图说明
[0046]
图1为本发明实施例1制备的式(9)所示的单体的核磁谱图;
[0047]
图2为本发明实施例2制备的式(11)所示的聚合物的核磁谱图;
[0048]
图3为本发明实施例3制备的式(13)所示的聚合物的核磁谱图;
[0049]
图4为本发明实施例4制备的式(15)所示的聚合物的核磁谱图;
[0050]
图5为本发明实施例5制备的聚芳醚酮中膦酰胺脱落率随时间变化曲线图;
[0051]
图6为本发明对比例1制备的聚醚醚酮中苯胺脱落率随时间变化曲线图。
具体实施方式
[0052]
为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制。
[0053]
鉴于现有技术中制备聚芳醚酮类聚合物的前驱体水解速度慢,而影响其加工和应用的问题,本技术从前驱体聚合物的结构设计角度出发,提供了一种新型的膦酰亚胺型前驱体聚合物,其实现了聚芳醚酮类聚合物的助溶以及助溶侧基快速水解脱落,实现了paek类聚合物更灵活的加工与应用。具体的,本发明实施例首先公开了如式(ⅰ)所示的膦酰亚胺缩聚单体,
[0054][0055]
其中,x选自卤素;
[0056]
r1和r2独立的选自氰基、取代或未取代的c1~c10的烷基,取代或未取代的c1~c5的烯基、取代或未取代的c6~c20的芳基。
[0057]
在本技术中,所述膦酰亚胺缩聚单体中,所述x优选f或cl;所述r1和r2独立的选自取代或未取代的c1~c6的烷基,其中取代的基团可以为乙烯基,取代或未取代的c6~c12的芳基,其中取代的基团可以为甲基,更具体地,所述r1和r2独立的选自以下并不限于以下基团中的一种:
[0058][0059][0060]
本技术上述膦酰亚胺缩聚单体是可以参与缩聚的膦酰亚胺单体。
[0061]
本技术还提供了如式(ⅰ)所示的膦酰亚胺缩聚单体的制备方法,包括以下步骤:
[0062]
a)将如式(ii)所示的4,4
’‑
二卤二苯酮、盐酸羟胺和醋酸钠在有机溶剂中混合,加热回流,得到如式(ⅲ)所示的4,4
’‑
二卤二苯酮肟;
[0063]
b)将所述4,4
’‑
二卤二苯酮肟和如式(ⅳ)所示的卤化膦化合物在弱碱环境中反应,得到如式(ⅰ)所示的膦酰亚胺缩聚单体;
[0064][0065]
其中,x选自卤素;
[0066]
r1和r2独立的选自氰基、取代或未取代的c1~c10的烷基,取代或未取代的c1~c5的烯基、取代或未取代的c6~c20的芳基。
[0067]
在上述制备过程中,在步骤a)中,所述4,4
’‑
二卤二苯酮和盐酸羟胺的摩尔比为1:1.1~1:1.6,所述4,4
’‑
二卤二苯酮与所述醋酸钠的摩尔比为1:1.6~1:2;所述加热回流的
时间为12~36h;所述有机溶剂为体积比为(6~1):1的乙醇和水的混合溶剂。上述步骤的具体反应式如下所示:
[0068][0069]
在步骤b)中,所述4,4
’‑
二卤二苯酮肟和所述弱碱的摩尔比为1:(1.1~1.5),所述反应的温度为-80~0℃,所述弱碱选自三乙胺、三甲胺、n,n-二异丙基乙胺等常见有机弱碱中的一种或几种;更具体地,所述4,4
’‑
二卤二苯酮肟和所述弱碱的摩尔比为1:(1.1~1.2),所述反应的温度为-35~0℃。上述反应的溶剂选自二氯甲烷、乙酸乙酯和乙腈中的一种或多种,具体的,所述反应的溶剂选自二氯甲烷。上述反应的反应式具体如下所示:
[0070][0071]
进一步的,本技术还提供了由上述膦酰亚胺缩聚单体制备的膦酰亚胺型前驱体聚合物,得到如式(
ⅴ
)所示的聚合物,
[0072][0073]
其中,r1和r2独立的选自氰基、取代或未取代的c1~c10的烷基,取代或未取代的c1~c5的烯基、取代或未取代的c6~c20的芳基;
[0074]
r3选自取代或未取代的c5~c30的芳基或杂环芳基。
[0075]
更具体地,所述r3选自以下并不限于以下基团中的一种:
[0076][0077][0078]
本技术还提供了如式(
ⅴ
)所示的膦酰亚胺型前驱体聚合物的制备方法,包括以下步骤:
[0079]
将如式(ⅵ)所示的二酚单体的醇溶液与氢氧化钠混合,反应,得到酚钠盐;
[0080]
将酚钠盐和如式(ⅰ)所示的膦酰亚胺缩聚单体反应,得到如式(
ⅴ
)所示的膦酰亚
胺型前驱体聚合物;
[0081]
go-r
3-oh(ⅵ);
[0082][0083]
其中,x选自卤素;
[0084]
r1和r2独立的选自氰基、取代或未取代的c1~c10的烷基,取代或未取代的c1~c5的烯基、取代或未取代的c6~c20的芳基;
[0085]
r3选自取代或未取代的c5~c30的芳基或杂环芳基;
[0086]
n为聚合度,优选10~5000。
[0087]
按照本发明,所述膦酰亚胺型前驱体聚合物的制备方法更具体为:
[0088]
a)将二酚单体的醇溶液与强极性非质子溶剂混合,在氮气保护下加入氢氧化钠固体,反应,得到酚钠盐;
[0089]
b)将步骤a)得到的反应体系加热至80~100℃以去除醇,再加入甲苯,加热至140~160℃,共沸除水;然后加入膦酰亚胺缩聚单体,加热至150~200℃,反应后得到膦酰亚胺型前驱体聚合物。
[0090]
在上述制备方法中,步骤a)中,所述强极性非质子溶剂选自n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜、n-甲基吡咯烷酮、二苯砜和环丁砜中的一种或多种;所述醇溶液选自甲醇、乙醇和异丙醇中的一种或多种。所述二酚单体和所述膦酰亚胺缩聚单体的摩尔比为1:1。上述制备膦酰亚胺型前驱体聚合物的反应式具体为:
[0091][0092]
在得到上述膦酰亚胺型前驱体聚合物之后,则将其在酸性条件下水解脱除膦酰胺侧基,以得到如式(ⅶ)所示的聚芳醚酮,具体为:
[0093]
将如式(
ⅴ
)所示的膦酰亚胺型前驱体聚合物与酸性溶液混合,水解,得到如式(ⅶ)所示的聚芳醚酮;
[0094][0095]
其中,r1和r2独立的选自氰基、取代或未取代的c1~c10的烷基,取代或未取代的c1~c5的烯基、取代或未取代的c6~c20的芳基;
[0096]
r3选自取代或未取代的c5~c30的芳基或杂环芳基;
[0097]
n为聚合度,优选10~5000。
[0098]
在制备聚芳醚酮的过程中,所述膦酰亚胺型前驱体聚合物的形态可以为丝状、粉状、块状或膜形态,对此本技术没有特别的限制。膦酰胺在水中溶解度较差,为了利于水解脱落的磷酰胺溶解在酸液中,所述酸溶液中可加入有机溶剂,所述酸溶液中有机溶剂与水的质量比为10:1~1:10,具体的,所述有机溶剂与水的质量比为3:1~1:5。所述有机溶剂可选自与水混溶的常见有机溶剂,具体可选自甲醇、乙醇、异丙醇、丙酮、丁酮、乙腈、四氢呋喃、二氧六环、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、二甲基亚砜和n-甲基吡咯烷酮中的一种或多种;所述酸液中的算可选自盐酸、硫酸、醋酸或磷酸。所述聚芳醚酮的制备过程具体为:
[0099][0100]
在膦酰亚胺缩聚单体的基础上,本技术提供了一种膦酰亚胺型前驱体聚合物,最终由其制备了聚芳醚酮类聚合物;相比于苯胺席夫碱,膦酰胺席夫碱由于膦酰胺极性大,所以该亚胺基团易被进攻水解,得到的前驱体聚合物可在弱酸且相对低的温度下快速水解,省去了长时间的酸煮过程,由此可更加灵活地制备聚芳醚酮类聚合物。
[0101]
为了进一步理解本发明,下面结合实施例对本发明提供的膦酰亚胺缩聚单体、其制备方法、膦酰亚胺型前驱体聚合物、其制备方法与聚芳醚酮的制备方法进行详细说明,本发明的保护范围不受以下实施例的限制。
[0102]
实施例1式(9)所示单体结构的合成
[0103]
将33g(0.1415mol)如(8)所示的结构加入500ml带有氮气进气口、机械搅拌与恒压滴液漏斗的三口烧瓶中,向瓶中加入250ml二氯甲烷作为溶剂,打开机械搅拌,接通氮气,将烧瓶置于-35℃冰乙醇浴中;待体系温度稳定后,向体系中加入21.63ml(0.1557mol)三乙胺,并向恒压滴液漏斗中加入30.71ml(0.1698mol)二苯基氯化膦;将二苯基氯化膦缓缓滴
入反应体系中,待全部滴入完毕后,反应1h后将反应恢复至室温;加入水淬灭未反应完的二苯基氯化膦,并用水洗涤有机体系,将有机体系重结晶,则得到最终产品n-(4,4
’‑
二氟二苯基亚甲基)-p,p-二苯基膦酰胺,如式(9)所示结构的单体,式(9)所示结构核磁谱图如图1所示。上述反应具体如下所示:
[0104][0105]
实施例2式(9)所示单体结构与式(10)所示单体结构缩聚为式(11)所示聚合物结构
[0106]
将6.6066g(0.06mol)式(10)所示结构的化合物加入500ml的带有氮气进气口、机械搅拌、分水器以及柱形冷凝管的三口烧瓶中,加入溶剂70ml二甲基亚砜与适量甲醇(50ml)溶解分散;搭建反应装置,并接通氮气,吹尽体系中的空气后,在氮气流下加入5.0g naoh(纯度96%,0.12mol)固体,打开机械搅拌,充分反应制备二酚钠盐溶液;打开油浴锅加热至体系温度为110℃,结合氮气将体系中的甲醇吹至分水器中,将分水器中的甲醇放干,向体系中加入30ml甲苯将分水器中装满甲苯,将温度升至150℃,利用甲苯共沸除去体系中的水与残余的甲醇,确保水分被充分除去后,将体系中甲苯放干吹出,加入25.046g(0.06mol)如式(9)所示单体;温度提升至160℃,聚合6h后,得到式(11)所示结构聚合物,式(11)所示结构聚合物的核磁谱图如图2所示。上述制备具体过程如下:
[0107][0108]
实施例3式(9)所示单体结构与式(12)所示单体结构缩聚为式(13)所示聚合物结构
[0109]
将12.8530g(0.06mol)式(12)所示结构的化合物加入500ml的带有氮气进气口、机械搅拌、分水器以及柱形冷凝管的三口烧瓶中,加入溶剂70ml二甲基亚砜与适量甲醇(50ml)溶解分散,搭建反应装置,并接通氮气,吹尽体系中的空气后,在氮气流下加入5.0g naoh(纯度96%,0.12mol)固体,打开机械搅拌,充分反应制备二酚钠盐溶液;打开油浴锅加
热至体系温度为110℃,结合氮气将体系中的甲醇吹至分水器中,将分水器中的甲醇放干,向体系中加入30ml甲苯将分水器中装满甲苯,将温度升至150℃,利用甲苯共沸除去体系中的水与残余的甲醇;确保水分被充分除去后,将体系中甲苯放干吹出,加入25.046g(0.06mol)式(9)所示结构的单体;温度提升至160℃,聚合6h后,得到式(13)所示结构聚合物,式(13)所示结构聚合物核磁谱图如图(3)所示,具体过程如下:
[0110][0111]
实施例4式(9)所示单体结构与式(14)所示单体结构缩聚为式(15)所示聚合物结构
[0112]
将13.6974g(0.06mol)式(14)所示结构的化合物加入500ml的带有氮气进气口、机械搅拌、分水器以及柱形冷凝管的三口烧瓶中,加入溶剂70ml二甲基亚砜与适量甲醇(50ml)溶解分散,搭建反应装置,并接通氮气,吹尽体系中的空气后,在氮气流下加入5.0g naoh(纯度96%,0.12mol)固体,打开机械搅拌,充分反应制备二酚钠盐溶液;
[0113]
打开油浴锅加热至体系温度为110℃,结合氮气将体系中的甲醇吹至分水器中,将分水器中的甲醇放干。向体系中加入30ml甲苯将分水器中装满甲苯,将温度升至150℃,利用甲苯共沸除去体系中的水与残余的甲醇,确保水分被充分除去后,将体系中甲苯放干吹出,加入25.046g(0.06mol)式(9)所示结构单体,温度提升至160℃,聚合6h后,得到式(15)所示结构聚合物,式(15)所示聚合物核磁谱图如图(4)所示,具体过程如下:
[0114][0115]
实施例5式(11)所示结构聚合物的酸性条件水解
[0116]
将式(11)所示结构的聚合物丝投入60℃的1mol/l的水:乙醇=3:1的盐酸溶液,反
应后得到聚芳醚酮,具体反应过程如下所示;通过紫外分光光度计检测溶液中侧基含量变化确定侧基脱落效果;
[0117][0118]
通过测试酸液中不同时间条件下式(16)所示结构的紫外吸收强度变化,确定侧基脱落率与比例,如图5所示。
[0119]
对比例1
[0120]
根据专利cn 110052178 a所述的内容,制备苯亚胺侧基修饰的聚醚醚酮(peekt),反应过程具体如下所示;将peekt制成丝状,运用同样的酸度条件进行测试;
[0121][0122]
通过测试酸液中式(17)所示结构的紫外吸收强度变化,确定侧基脱落率与比例,结果如图6所示。
[0123]
由图5和图6可知,式(11)所示聚合物的膦酰胺侧基脱落速率要远远高于对比例中peekt的苯胺侧基脱落速率。
[0124]
以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。
[0125]
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。