1.本发明涉及化学合成技术领域,具体涉及一种特格拉赞中间体的制备方法。
背景技术:
2.特格拉赞,也叫特戈拉赞(tegoprazan),其化学结构式如下:
[0003][0004]
特格拉赞(tegoprazan)是一种竞争性钾离子酸阻滞剂(p-cab)和氢离子钾离子交换atp酶(h /k atpase)抑制剂,批准用于治疗胃食管反流病及糜烂性食管炎。特格拉赞最初由辉瑞制药研发,后在2008年授权给raqualia pharma合作开发。2018年7月获韩国食品药品安全部(mfds)批准上市,由cj healthcare在韩国上市销售。
[0005]
胃食管反流病是一种十分常见的消化道疾病,在人群中发病率很高,主要症状表现是烧心、胸痛、泛酸和反流等,严重时会导致食管炎。调查显示,大约有7%的人几乎每天发生反流症状。目前治疗该类疾病的方法由药物治疗和手术治疗,但都有各自的缺点,所以有必要研制新的治疗该类常见疾病的药物。tegoprazan是一种钾竞争性酸阻断剂,被认为是目前治疗胃食管反流疾病最先进的药物,因为质子泵抑制剂是最常见的治疗胃食管反流病的药物,而tegoprazan恰好能克服质子泵抑制剂的缺点。tegporazan为该类疾病的治疗提供了新的选择,并且一定程度弥补了其他药物的缺点。
[0006]
目前,关于合成特格拉赞中间体方法的报道主要有以下几条:
[0007]
路线一,中国发明专利cn101341149b中公开了下述合成路线,参见式1:
[0008][0009]
该路线中,n-{4-溴-2-硝基-6-[(苯甲基)氧基]苯基}乙酰胺制备n-{4-氰基-2-硝基-6-[(苯甲基)氧基]苯基}乙酰胺的步骤中需要用到的金属氰化物试剂为氰化锌,氰化锌是一种极高毒性的原料,危险性大,易对环境造成长期污染,不利于放大生产。
[0010]
路线二:公开号为w02008114123的发明专利公开了下述合成路线,参见式2:
[0011][0012]
该路线除了使用到贵金属pd催化剂,还需要用到co于压力釜中反应,成本高,且co有毒,存在一定的危险性。
[0013]
路线三:公开号为cn101341149b的发明专利申请公开了下述合成路线,参见式3:
[0014][0015]
该路线使用到nbs溴代物,由于羟基用苄基先保护起来,因此,该步反应存在较多副反应,导致目标产物收率偏低。
[0016]
路线四:公开号为cn114249694a的发明专利申请公开了下述合成路线,参见式4:
[0017][0018]
该路线中的环和步骤使用到氯代丁二酰亚胺,由于羟基用苄基先保护起来,因此,同路线三一样,该步反应存在较多副反应,导致目标产物收率偏低。
[0019]
基于上述现有技术中特格拉赞中间体合成路线中存在的问题,本发明提出一种特格拉赞中间体的制备方法,用于制备下式5中的特格拉赞中间体,以期解决上述问题。
[0020]
技术实现要素:
[0021]
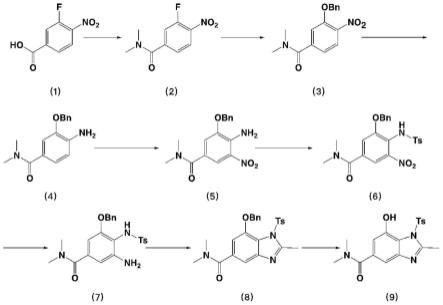
本发明的目的在于,克服现有技术中存在的缺陷,提供一种特格拉赞中间体的制备方法,以3-氟-4-硝基苯甲酸(化合物1)为起始原料,依次经过缩合、取代、还原、硝化、氨基保护、还原、环和、脱保护基,最终得到目标产物化合物9,反应路线中未使用到贵重金属,成本较低,各步反应步骤均操作安全简单,环境污染小,后处理简单,工业化生产可行性高;与背景技术中反应路线四相比较,本发明的一种特格拉赞中间体的制备方法,首先,在步骤s2中,化合物2与苄醇反应得到具有羟基保护作用的化合物3,苄醇相对于溴化苄温和性好,适合放大生产;其次,步骤s7中的环合反应操作简单,更有利于工业化生产。
[0022]
为实现上述目的,本发明的技术方案是设计一种特格拉赞中间体的制备方法,其合成路线如下式6所示,
[0023][0024]
包括如下步骤:
[0025]
s1:化合物1与二甲胺盐酸盐在酰胺化试剂条件下,反应制备得到化合物2;
[0026]
s2:化合物2在碱性条件下,与苄醇反应制备得到化合物3;
[0027]
s3:化合物3经还原反应制备得到化合物4;
[0028]
s4:化合物4经硝化反应制备得到化合物5;
[0029]
s5:化合物5经氨基保护得到化合物6;
[0030]
s6:化合物6经还原反应制备得到化合物7;
[0031]
s7:化合物7经环和反应制备得到化合物8;
[0032]
s8:化合物8经脱保护基反应制备得到特格拉赞中间体化合物9。
[0033]
优选的技术方案是,所述的步骤s1具体操作为:以化合物1和二甲胺盐酸盐为原料,于碱、酰胺化试剂存在的溶剂中反应,制备得到化合物2,其中化合物1与二甲胺盐酸盐、碱、酰胺化试剂的摩尔投料比为1:1:2:1,化合物1与溶剂的质量投料比为1:2~3,反应温度为0~30℃,反应时间为1~2h。
[0034]
进一步优选的技术方案有,所述的步骤s1中:所用的酰胺化试剂为1,1'-羰基二咪唑、六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺、n,n-二异丙基碳二亚胺、二环己基甲烷二亚胺和1-丙基磷酸环酐中的一种,所用的碱为n,n-二异丙基乙胺或三乙胺,所用的溶剂为二氯甲烷、n,n-二甲基甲酰胺、乙腈、四氢呋喃和乙酸乙酯中的一种,反应温度为20~30℃,反应时间为1~2h。
[0035]
优选的技术方案还有,所述的步骤s2中,所用的碱为氢化钠、叔丁醇钾、乙醇钠中的一种,所用的溶剂为四氢呋喃、n,n-二甲基甲酰胺中的一种,化合物2与苄醇、碱、溶剂的质量投料比为1:0.5~1:0.2~0.6:6~8,反应温度为0~30℃,反应时间为0.5~1h。
[0036]
优选的技术方案还有,所述的步骤s3中,所用的还原剂为还原铁粉、锌粉、连二亚硫酸钠、氯化亚锡、水合肼中的一种,所用的溶剂为甲醇-水、乙醇-水、异丙醇-水混合体系中的一种,化合物3与还原剂、溶剂的质量投料比为1:2~3:6~8,反应温度为50~80℃,反应时间为4~6h。
[0037]
优选的技术方案还有,所述的步骤s4中,所用的硝化试剂为浓硫酸-硝酸钾、浓硫酸-硝酸钠、浓硝酸-浓硫酸、浓硝酸-冰醋酸混合体系中的一种,化合物4与硝化试剂的质量投料比为1:2~3,反应温度为25~35℃,反应时间为3~4h。
[0038]
优选的技术方案还有,所述的步骤s5中,氨基保护所用的保护剂为对甲苯磺酰基、苄基、叔丁氧羰基、对甲氧基苄基中的一种,所用的碱为n,n-二异丙基乙胺、三乙胺中的一种,所用的溶剂为二氯甲烷,化合物5与保护剂、碱的摩尔投料比为1:1~1.5:2~3,化合物5与溶剂的质量投料比为1:8~10,反应过程为0~5℃反应0.5h,然后升温至25~35℃,反应时间为2h。
[0039]
优选的技术方案还有,所述的步骤s6中,所用的还原剂为还原铁粉、锌粉、连二亚硫酸钠、氯化亚锡、水合肼中的一种,所用的溶剂为甲醇-水、乙醇-水、异丙醇-水混合体系中的一种,化合物6与还原剂的摩尔投料比为1:4~5,化合物6与溶剂的质量投料比为1:6~8,反应温度为50~80℃,反应时间为4~6h。
[0040]
优选的技术方案还有,所述的步骤s7中,所用的环合试剂为原乙酸三乙酯,化合物7与环合试剂摩尔投料比为1:2,所用的溶剂为甲苯、乙苯中的一种,化合物7与溶剂的质量投料比为1:5~7。
[0041]
优选的技术方案还有,所述的步骤s8中,所用的脱保护剂为10%pd/c和甲酸铵,所用的溶剂为甲醇、乙醇、异丙醇中的一种,化合物8与10%pd/c、溶剂的质量投料比为1:0.2~0.3:15~20,化合物8与甲酸铵的摩尔投料比为1:5。
[0042]
本发明的优点和有益效果在于:
[0043]
1、本发明的一种特格拉赞中间体的制备方法,以3-氟-4-硝基苯甲酸(化合物1)为起始原料,依次经过缩合、取代、还原、硝化、氨基保护、还原、环合和脱保护基,最终得到目
标产物化合物9,反应路线中,使用的起始物料和辅料都是常规的大宗化工原料,便宜易得;各步反应步骤均操作安全简单,环境污染小,后处理简单,工业化生产可行性高。
[0044]
2、背景技术中反应路线四中,首先,羟基保护是用溴化苄与酚羟基反应得到,其中溴化苄使用过程中会产生溴化氢,有很强的刺激性,不便于放大生产;其次,环合反应操作步骤繁琐,不便于工业化生产。本发明的一种特格拉赞中间体的制备方法,首先,在步骤s2中,化合物2与苄醇反应得到具有羟基保护作用的化合物3,苄醇使用过程中不会产生刺激性的溴化氢,相对于溴化苄绿色环保,温和性好,适合放大生产;其次,路线四中还用到了醋酐,该产品属于管制品类-易制毒化学品品种名录第二类,不得非法销售或购买,购买较繁琐。步骤s7中的环合使用的试剂则不存在这样的问题,同时,环合产生的副产物为乙醇,绿色清洁,且反应操作简单,更有利于工业化生产。
附图说明
[0045]
图1是本发明一种特格拉赞中间体的制备方法的合成反应路线;
[0046]
图2是实施例1中步骤s1所制备化合物2的核磁图谱;
[0047]
图3是实施例1中步骤s2所制备化合物3的核磁图谱;
[0048]
图4是实施例1中步骤s3所制备化合物4的核磁图谱;
[0049]
图5是实施例1中步骤s4所制备化合物5的核磁图谱;
[0050]
图6是实施例1中步骤s5所制备化合物6的核磁图谱;
[0051]
图7是实施例1中步骤s6所制备化合物7的核磁图谱;
[0052]
图8是实施例1中步骤s7所制备化合物8的核磁图谱;
[0053]
图9是实施例1中步骤s8所制备化合物9的核磁图谱。
具体实施方式
[0054]
下面结合附图和实施例,对本发明的具体实施方式作进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不能以此来限制本发明的保护范围。
[0055]
实施例1
[0056]
如图1所示,采用本发明的方法制备特格拉赞中间体化合物9,包括如下操作步骤:
[0057]
s1:化合物2的制备,反应式参见下7:
[0058][0059]
室温下,250ml反应瓶中依次投入二氯甲烷(dcm)(20ml)、化合物1(10g,54.02mmol)、1,1'-羰基二咪唑(cdi)(8.76g,54.02mmol)、n,n-二异丙基乙胺(diea)(13.94g,108.04mmol)和二甲胺盐酸盐(4.40g,54.02mmol)氮气保护下,开启搅拌,20~30℃保温反应1h。保温结束后,薄层色谱法(tlc)监控反应进程至几乎无化合物1残留,停止反应。将反应液倒入饱和碳酸氢钠溶液(200ml),室温搅拌1h。搅拌结束后,分出水层用dcm
(20ml
×
2)萃取。合并有机层,用无水硫酸钠干燥,减压浓缩至干得到10.85g淡黄色固体(化合物2),收率94.76%,其核磁图谱参见附图2,核磁数据为:1h nmr(400mhz,cdcl3)δ8.10(dd,j=7.2hz,8.4hz,1h),7.35(m,2h),3.12(s,3h),2.97(s,3h).ms(esi):m/z 213[m h]
;
[0060]
s2:化合物3的制备,反应式参见下式8:
[0061][0062]
室温下,150ml反应瓶中投入60%nah(5.66g,141.5mmol)和n,n-二甲基甲酰胺(dmf)(50ml),开启搅拌和降温。内温降至0~10℃,保温搅拌5min。保温结束后,向反应液中依次投入苄醇(5.60g,51.78mmol)和化合物(1)/dmf(10.00g/25ml,47.17mmol),20~30℃保温反应1h。保温结束后,tlc监控反应进程至无原料残留,停止反应。反应液倒入水中(400ml),室温搅拌10min,用dcm(50ml
×
3)萃取。合并有机层,依次用饱和氯化铵(50ml)和水中(50ml)洗涤,无水硫酸钠干燥,减压浓缩至干得到14.05g油状物(化合物3),收率99.29%,其核磁图谱参见附图3,核磁数据为:1h nmr(400mhz,cdcl3)δ2.96(s,3h),3.09(s,3h),5.25(s,2h),7.04(d,2h,j=8.4hz),7.16(s,1h),7.40(m,5h),7.87(d,1h,j=8.0hz)。ms(esi):m/z 301[m h]
;
[0063]
s3:化合物4的制备,反应式参见下式9:
[0064][0065]
室温下,反应瓶中依次投入95%etoh/h2o(80ml)和化合物3(10.00g,33.33mmol),开启加热和搅拌。内温升至50~60℃,分批投入na2s2o4(29.00g,166.67mmol),大约10min投毕。升温至回流(78℃),保温反应5h。保温结束后,tlc中控无原料残留,停止反应。将反应液投入水中(200ml)搅拌析晶1h。析晶结束后,过滤,滤饼50℃烘干得8g(化合物4),收率88.89%,其核磁图谱参见附图4,核磁数据为:1h nmr(400mhz,cdcl3)δ3.01(s,6h),4.02(s,2h),5.09(s,2h),6.68(d,1h,j=8.0hz),6.96(d,1h,j=8.0hz),7.01(s,1h),7.33(m,5h)。ms(esi):m/z 271[m h]
;
[0066]
s4:化合物5的制备,反应式参见下式10:
[0067][0068]
室温下,反应瓶中投入浓硫酸(50ml),开启搅拌和降温,待反应瓶内温度降至0~5℃,投入化合物4(10.00g,37.03mmol),保持反应瓶内温度10℃以下搅拌直至化合物4完全溶解。分批投入硝酸钾(20.00g,197.82mmol),控制反应瓶内温度10℃以下。硝酸钾投毕后,升至25~35℃保温反应3h。保温结束后,tlc监控反应进程至无原料残留,停止反应。将反应液缓慢倒入冰水中(500g),控制温度不超过10℃。待转料结束后,用氢氧化钠固体调节ph值7~8。过滤,滤饼用20ml水漂洗,滤饼50℃烘干得10.36g棕色固体(化合物5),收率88.70%,其核磁图谱参见附图5,核磁数据为:1h nmr(500mhz,cdcl3)δ12.27(d,j=2.2hz,1h),11.83~11.34(m,6h),10.59(dd,j=61.2,8.2hz,2h),9.42(t,j=1.0hz,2h),7.26(s,6h)。(esi):m/z316[m h]
;
[0069]
s5:化合物6的制备,反应式参见下式11:
[0070][0071]
室温下,反应瓶中依次投入dcm(60ml)、化合物5(10.00g,31.75mmol)和tea(6.41g,63.47mmol),开启搅拌降温。内温降至0~5℃,滴加对甲苯磺酰氯/dcm(7.25g/20ml,38.03mmol)溶液。滴加过程中控制内温不超过5℃,约30min滴毕。先0~5℃保温反应30min,再升高至25~35℃,保温反应2h。保温结束后,tlc中控无原料残留,停止反应。反应液用dcm(200ml)稀释,依次用饱和碳酸氢钠(50ml)、水(50ml)和盐水(50ml)洗涤。最后用无水硫酸钠干燥,减压浓缩至干得到13.52g淡黄色固体(化合物6),收率90%,其核磁图谱参见附图6,核磁数据为:1h nmr(500mhz,cdcl3)δ13.61(s,1h),12.63(d,j=2.0hz,1h),12.17~11.92(m,2h),11.75~11.41(m,8h),9.44(t,j=1.0hz,2h),7.26(s,6h),6.67(d,j=0.9hz,3h).ms(esi):m/z 471[m h]
;
[0072]
s6:化合物7的制备,反应式参见下式12:
[0073][0074]
室温下,反应瓶中依次投入95%etoh/h2o(100ml)和化合物6(10.00g,
21.28mmol),开启加热和搅拌。内温升至50~60℃,分批投入na2s2o4(18.53g,93.71mmol),大约10min投毕。升温至回流(78℃),保温反应5h。保温结束后,tlc中控无原料残留,停止反应。将反应液投入水中(200ml)搅拌析晶1h。析晶结束后,过滤,滤饼50℃烘干得8.61g(化合物7),收率91%,其核磁图谱参见附图7,核磁数据为:1h nmr(500mhz,cdcl3)δ13.44(s,1h),12.10~11.90(m,2h),11.76~11.46(m,7h),11.36~10.91(m,2h),10.17(dd,j=56.2,7.1hz,2h),9.45(t,j=1.0hz,2h),7.26(s,6h)6.67(d,j=0.9hz,3h).ms(esi):m/z 441[m h]
;
[0075]
s7:化合物8的制备,反应式参见下式13:
[0076][0077]
室温下,反应瓶中依次投入甲苯(80ml)、原乙酸三乙酯(7.38g,45.50mmol)、化合物7(10.00g,22.75mmol)和浓盐酸(2滴),开启加热和搅拌。升温至80℃,保温反应1h。保温结束后,tlc监控反应进程至无原料残留,停止反应,搅拌降温。内温降至室温,反应液用dcm(200ml)稀释,依次用饱和碳酸氢钠(50ml)、水(50ml)和饱和盐水(50ml)洗涤。最后用无水硫酸钠干燥,减压浓缩至干得到类白色固体10.23g(化合物8),收率96.68%,其核磁图谱参见附图8,核磁数据为:1h nmr(400mhz,cdcl3)δ2.40(s,3h),2.81(s,3h),2.87(s,3h),3.11(s,3h),5.32(s,2h),6.86(s,1h),7.28~7.36(m,5h),7.43(s,1h),7.45(s,1h),7.69(s,1h),7.78(d,2h,j=8.4hz)。ms(esi):m/z 464[m h]
;
[0078]
s8:化合物9的制备,反应式参见下式14:
[0079][0080]
室温下,反应瓶中依次投入异丙醇(200ml)、化合物8(10.00g,21.57mmol)、10%pd/c(2.0g)和甲酸铵(6.82g,107.85mmol),开启加热和搅拌。升温至回流,保温反应30min。保温结束后,tlc监控反应进程至无原料残留,停止反应。趁热过滤,滤饼用预热的异丙醇(20ml
×
3)洗涤至无产品。滤液减压浓缩至干。浓缩物用10%meoh/dcm(50ml)溶解,饱和盐水洗涤,无水硫酸钠干燥,减压浓缩至干得到7.98g白色固体(化合物9),收率99.06%,其核磁图谱参见附图9,核磁数据为:1h nmr(400mhz,cdcl3)δ2.41(s,3h),2.79(s,3h),3.02(s,3h),3.15(s,3h),6.92(s,1h),7.32(d,2h,j=8.1hz),7.63(s,1h),7.82(d,2h,j=8.1hz)。ms(esi):m/z 374[m h]
。
[0081]
实施例2
[0082]
如图1所示,采用本发明的方法制备特格拉赞中间体化合物9,包括如下操作步骤:
[0083]
s1:化合物2的制备,反应式参见下7:
[0084][0085][0086]
室温下,100ml反应瓶中依次投入n,n-二甲基甲酰胺(dmf)(20ml)、化合物(1)(10.00g,54.02mmol)、六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷(pybop)(28.11g,54.02mmol)、三乙胺(tea)(10.92g,108.04mmol)和二甲胺盐酸盐(4.40g,54.02mmol),开启搅拌降温,内温降至0~10℃保温反应2h。保温结束后,薄层色谱法(tlc)监控反应进程至几乎无化合物(1)残留,停止反应。将反应液倒入50%乙醇水溶液(200ml)中,室温搅拌1h。搅拌结束后,过滤,滤饼用水(20ml)室温打浆洗涤,过滤,抽干,50℃鼓风干燥得11.30g淡黄色固体(化合物2),收率98.58%,其核磁数据为:1h nmr(500mhz,cdcl3)δ7.25(dd,j=8.6,5.0hz,1h),6.89(dd,j=8.5,1.9hz,1h),6.85(dd,j=8.0,1.9hz,1h),2.08(s,6h).ms(esi):m/z 213[m h]
;
[0087]
s2:化合物3的制备,反应式参见下式8:
[0088][0089]
室温下,150ml反应瓶中投入叔丁醇钾(5.50g,49.11mmol)和四氢呋喃(thf)(60ml),开启搅拌和降温。内温降至0~10℃,保温搅拌5min。保温结束后,向反应液中依次投入苄醇(5.60g,51.85mmol)和化合物(2)/thf(10.00g/15ml,47.17mmol),20~30℃保温反应1h。保温结束后,tlc监控反应进程至无原料残留,停止反应。反应液倒入水中(400ml),室温搅拌10min,用dcm(50ml
×
3)萃取。合并有机层,依次用饱和氯化铵(50ml)和水中(50ml)洗涤,无水硫酸钠干燥,减压浓缩至干得到13.98油状物(化合物3),收率98.79%,其核磁数据为:1h nmr(500mhz,cdcl3)δ8.06(d,j=2.0hz,1h),8.05(d,j=1.8hz,1h),7.73(d,j=1.9hz,2h),7.72(d,j=5.9hz,4h),7.64(t,j=6.7hz,4h),7.59(d,j=6.4hz,2h),5.53(t,j=1.0hz,4h),3.29(s,11h);化合物3的质谱数据为:ms(esi):m/z 301[m h]
,与理论质谱数据一致。
[0090]
s3:化合物4的制备,反应式参见下式9:
[0091][0092]
室温下,反应瓶中依次投入50%meoh/h2o(80ml)/化合物3(10.00g,33.33mmol)、铁粉(25.08g,44.79mmol)和浓盐酸(0.5ml),开启加热和搅拌。升至回流(65℃),保温反应5h。保温结束后,tlc监控无化合物(3)残留,反应液用硅藻土过滤,滤液减压浓缩至干。上述浓缩物用5%meoh/dcm溶解,饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥。过滤后减压浓缩至干得到8.50g产物(化合物4),收率94.44%,其核磁数据为:1h nmr(500mhz,cdcl3)δ6.08(d,j=1.1hz,1h),6.06(d,j=1.2hz,1h),6.02(d,j=1.8hz,1h),6.00(d,j=1.0hz,2h),5.98(s,1h),5.93(s,1h),5.82(d,j=1.9hz,1h),5.46(d,j=8.6hz,1h),3.82(s,2h),2.68(d,j=7.1hz,1h),2.56(d,j=7.1hz,1h),1.65(s,7h).ms(esi):m/z 271[m h]
。
[0093]
s4:化合物5的制备,反应式参见下式10:
[0094][0095]
室温下,反应瓶中投入浓硫酸(60ml)和化合物(4)(10.00g,37.04mmol),开启搅拌和降温,待反应瓶内温度降至0~5℃,滴加85%浓硝酸(20.00g,350.79mmol),滴加过程中保持反应瓶内温度10℃以下,滴毕后,10℃以下保温1h。保温结束后,升温至25~35℃,保温反应3h。保温结束后,tlc监控无化合物(4)残留。将反应液缓慢滴入冰水(500g)水中,滴加过程保持水温度不超过10℃,有固体析出。滴加结束后,室温搅拌1h后过滤,滤饼用水洗至ph为7~8,滤饼50℃烘干得10.51g棕色固体(化合物5),收率90.00%,其核磁数据为:1h nmr(500mhz,cdcl3)δ7.26(d,j=2.2hz,1h),6.67(dd,j=7.1,1.2hz,2h),6.63~6.50(m,4h),5.64(s,1h),5.52(d,j=8.2hz,1h),4.41(s,2h),2.25(s,6h)。ms(esi):m/z 316[m h]
。
[0096]
s5:化合物6的制备,反应式参见下式11:
[0097][0098]
室温下,反应瓶中依次投入dcm(80ml)、化合物5(10.00g,31.75mmol)和diea
(12.29g,95.25mmol),开启搅拌降温。内温降至0~5℃,滴加对甲苯磺酰氯/dcm(15.04g/20ml,78.89mmol)溶液。滴加过程中控制内温不超过5℃,约30min滴毕。先0~5℃保温反应30min,再升高至25~35℃,保温反应2h。保温结束后,tlc中控无原料残留,停止反应。反应液用dcm(200ml)稀释,依次用饱和碳酸氢钠(50ml)、水(50ml)和盐水(50ml)洗涤。最后用无水硫酸钠干燥,减压浓缩至干得到13.52g淡黄色固体(化合物6),收率90%,其核磁数据为:1h nmr(500mhz,cdcl3)δ7.26(s,1h),6.28(d,j=2.0hz,1h),5.60(d,j=8.4hz,2h),5.34(d,j=2.2hz,2h),5.33(s,1h),5.26(d,j=1.2hz,1h),5.25(s,1h),5.23(s,2h),3.09(s,2h),0.91(s,6h),0.32(d,j=0.9hz,3h)。ms(esi):m/z 471[m h]
。
[0099]
s6:化合物7的制备,反应式参见下式12:
[0100][0101]
室温下,反应瓶中依次投入50%meoh/h2o(60ml)/化合物3(10.00g,21.28mmol)、铁粉(5.96g,106.4mmol)和浓盐酸(1ml),开启加热和搅拌。升至回流(65℃),保温反应4h。保温结束后,tlc监控无化合物(3)残留,反应液用硅藻土过滤,滤液减压浓缩至干。上述浓缩物用5%meoh/dcm溶解,饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥。过滤后减压浓缩至干得到9.2g产物(化合物7),收率97.23%,其核磁数据为:1h nmr(500mhz,cdcl3)δ13.44(s,1h),11.95(d,j=8.4hz,2h),11.68(dd,j=7.1,1.2hz,2h),11.61(t,j=6.7hz,2h),11.59~11.51(m,3h),11.21~11.17(m,2h),10.22(d,j=7.1hz,1h),10.11(d,j=7.1hz,1h),9.45(s,2h),7.26(s,6h),6.67(s,3h)。ms(esi):m/z 441[m h]
。
[0102]
s7:化合物8的制备,反应式参见下式13:
[0103][0104]
室温下,反应瓶中依次投入乙苯(60ml)、原乙酸三乙酯(7.38g,45.46mmol)、化合物7(10.00g,22.73mmol)和浓盐酸(2滴),开启加热和搅拌。升温至90℃,保温反应1h。保温结束后,tlc监控反应进程至无原料残留,停止反应,搅拌降温。内温降至室温,反应液用dcm(200ml)稀释,依次用饱和碳酸氢钠(50ml)、水(50ml)和饱和盐水(50ml)洗涤。最后用无水硫酸钠干燥,减压浓缩至干得到类白色固体9.82g(化合物8),收率93.32%,其核磁数据为:1h nmr(500mhz,cd4o)δ3.30~3.25(m,2h),3.01(dd,j=7.1,1.2hz,2h),2.97~2.92(m,3h),2.89(d,j=7.2hz,1h),2.87~2.82(m,2h),0.77(t,j=1.0hz,2h),-1.41(s,6h),-1.71(s,3h),-2.00(s,3h)。ms(esi):m/z 464[m h] ;
[0105]
s8:化合物9的制备,反应式参见下式14:
[0106][0107]
室温下,反应瓶中依次投入甲醇(200ml)、化合物8(10.00g,21.57mmol)、10%pd/c(2.00g)和甲酸铵(6.82g,107.85mmol),开启加热和搅拌。升温至回流,保温反应30min。保温结束后,tlc监控反应进程至无原料残留,停止反应。趁热过滤,滤饼用预热的甲醇(20ml
×
3)洗涤至无产品。滤液减压浓缩至干。浓缩物用10%meoh/dcm(50ml)溶解,饱和盐水洗涤,无水硫酸钠干燥,减压浓缩至干得到7.88g白色固体(化合物9),收率97.81%,其核磁数据为:1h nmr(500mhz,cdcl3)δ7.73(d,j=8.8hz,2h),7.67(d,j=2.2hz,1h),7.30~7.25(m,3h),3.03(s,6h),2.73(s,3h),2.43(s,3h)。ms(esi):m/z 374[m h]
。
[0108]
实施例3
[0109]
如图1所示,采用本发明的方法制备特格拉赞中间体化合物9,包括如下操作步骤:
[0110]
s1:化合物2的制备,反应式参见下7:
[0111][0112]
室温下,250ml反应瓶中依次投入乙腈(acn)(30ml)、化合物1(10.00g,54.02mmol)、n,n-二异丙基碳二亚胺(dic)(6.82g,54.02mmol),n,n-二异丙基乙胺(diea)(13.94g,108.04mmol)和二甲胺盐酸盐(4.40g,54.02mmol),开启搅拌降温。内温降至10~20℃保温反应2h。保温结束后,薄层色谱法(tlc)监控反应进程至几乎无化合物(1)残留,停止反应。将反应液倒入饱和碳酸氢钠溶液(200ml),室温搅1h。搅拌结束后,分出水层用dcm(20ml
×
2)萃取。合并有机层,用无水硫酸钠干燥,减压浓缩至干得到10.85g淡黄色固体(化合物2),收率96.51%,其核磁数据为:1h nmr(500mhz,cdcl3)δ7.25(dd,j=8.6,5.0hz,1h),6.85~6.89(dd,j=8.5,1.9hz,2h),2.08(s,6h)。ms(esi):m/z 213[m h]
;
[0113]
s2:化合物3的制备,反应式参见下式8:
[0114][0115]
室温下,150ml反应瓶中投入乙醇钠(6.00g,88.22mmol)和n,n-二甲基甲酰胺
(dmf)(50ml),开启搅拌和降温。内温降至0~10℃,保温搅拌5min。保温结束后,向反应液中依次投入苄醇(5.60g,52.33mmol)和化合物(1)/dmf(10.00g/15ml,47.17mmol),20~30℃保温反应1h。保温结束后,tlc监控反应进程至无原料残留,停止反应。反应液倒入水中(400ml),室温搅拌10min,用dcm(50ml
×
3)萃取。合并有机层,依次用饱和氯化铵(50ml)和水中(50ml)洗涤,无水硫酸钠干燥,减压浓缩至干得到14.01油状物(化合物3),收率99.00%,其核磁数据为:1h nmr(500mhz,cdcl3)δ8.06(d,j=2.0hz,1h),8.05(d,j=1.8hz,1h),7.73(d,j=1.9hz,2h),7.72(d,j=5.9hz,4h),7.64(t,j=6.7hz,4h),7.59(d,j=6.4hz,2h),5.53(t,j=1.0hz,4h),3.29(s,11h)。ms(esi):m/z 301[m h]
;
[0116]
s3:化合物4的制备,反应式参见下式9:
[0117][0118]
室温下,反应瓶中依次投入90%异丙醇/h2o(100ml)、化合物3(10.00g,33.33mmol)和氯化亚锡(20.01g,105.53mmol),开启加热和搅拌。升温至回流(80℃),保温反应5h。保温结束后,tlc中控无原料残留,停止反应。反应液用硅藻土过滤,滤液减压浓缩至干。上述浓缩物用5%meoh/dcm溶解,饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥。过滤后减压浓缩至干得到8.93g产物(化合物4),收率99.22%,其核磁数据为:1h nmr(500mhz,cdcl3)δ6.08(d,j=1.1hz,1h),6.06(d,j=1.2hz,1h),6.02(d,j=1.8hz,1h),6.00(d,j=1.0hz,2h),5.98(s,1h),5.93(s,1h),5.82(d,j=1.9hz,1h),5.46(d,j=8.6hz,1h),3.82(s,2h),2.68~2.56(d,j=7.1hz,2h),1.65(s,7h).ms(esi):m/z271[m h]
;
[0119]
s4:化合物5的制备,反应式参见下式10:
[0120][0121]
室温下,反应瓶中投入冰醋酸(60ml)和化合物(4)(10.00g,37.03mmol),开启搅拌和降温,待反应瓶内温度降至0~5℃,滴加85%浓硝酸(20.00g,350.79mmol),滴加过程中保持反应瓶内温度10℃以下,滴毕后,10℃以下保温1h。保温结束后,升温至25~35℃,保温反应4h。保温结束后,tlc监控无化合物(4)残留。将反应液缓慢滴入冰水(500g)水中,滴加过程保持水温度不超过10℃,有固体析出。滴加结束后,室温搅拌1h后过滤,滤饼用水洗至ph为7~8,滤饼50℃烘干得10.69g棕色固体(化合物5),收率91.63%,其核磁数据为:1h nmr(500mhz,cdcl3)δ7.26(d,j=2.2hz,1h),6.67(dd,j=7.1,1.2hz,2h),6.63~6.50(m,4h),5.64(s,1h),5.52(d,j=8.2hz,1h),4.41(s,2h),2.25(s,6h)。ms(esi):m/z 316[m h]
。
[0122]
s5:化合物6的制备,反应式参见下式11:
[0123][0124]
室温下,反应瓶中依次投入dcm(60ml)、化合物5(10.00g,31.75mmol)和tea(6.41g,63.50mmol),开启搅拌降温。内温降至0~5℃,滴加对甲苯磺酰氯/dcm(6.11g/20ml,32.05mmol)溶液。滴加过程中控制内温不超过5℃,约30min滴毕。先0~5℃保温反应30min,再升高至25~35℃,保温反应2h。保温结束后,tlc中控无原料残留,停止反应。反应液用dcm(200ml)稀释,依次用饱和碳酸氢钠(50ml)、水(50ml)和盐水(50ml)洗涤。最后用无水硫酸钠干燥,减压浓缩至干得到13.01g淡黄色固体(化合物6),收率86.54%,其核磁数据为:1h nmr(500mhz,cdcl3)δ7.26(s,1h),6.28(d,j=2.0hz,1h),5.60(d,j=8.4hz,2h),5.34(d,j=2.2hz,2h),5.33(s,1h),5.26(d,j=1.2hz,1h),5.25(s,1h),5.23(s,2h),5.19(t,j=1.4hz,0h),3.09(s,2h),0.91(s,6h),0.32(d,j=0.9hz,3h)。ms(esi):m/z 471[m h]
;
[0125]
s6:化合物7的制备,反应式参见下式12:
[0126][0127]
室温下,反应瓶中依次投入90%异丙醇/h2o(80ml)和化合物6(10.00g,21.28mmol),开启加热和搅拌。内温升至50~60℃,分批投入na2s2o4(22.22g,127.68mmol),大约10min投毕。升温至回流(78℃),保温反应5h。保温结束后,tlc中控无原料残留,停止反应。将反应液投入水中(200ml)搅拌析晶1h。析晶结束后,过滤,滤饼50℃烘干得8.61g(化合物7),收率91%,其核磁数据为:1h nmr(500mhz,cdcl3)δ13.44(s,1h),11.95(d,j=8.4hz,2h),11.68(dd,j=7.1,1.2hz,2h),11.61(t,j=6.7hz,2h),11.59~11.51(m,3h),11.21~11.17(m,2h),10.22(d,j=7.1hz,1h),10.11(d,j=7.1hz,1h),9.45(s,2h),7.26(s,6h),6.67(s,3h)。ms(esi):m/z 441[m h]
;
[0128]
s7:化合物8的制备,反应式参见下式13:
[0129][0130]
室温下,反应瓶中依次投入甲苯(70ml)、原乙酸三乙酯(7.38g,45.45mmol)、化合物7(10.00g,22.73mmol)和浓盐酸(2滴),开启加热和搅拌。升温至回流(110℃),保温反应
1h。保温结束后,tlc监控反应进程至无原料残留,停止反应,搅拌降温。内温降至室温,反应液用dcm(200ml)稀释,依次用饱和碳酸氢钠(50ml)、水(50ml)和饱和盐水(50ml)洗涤。最后用无水硫酸钠干燥,减压浓缩至干得到类白色固体10.58g(化合物8),收率99.98%,其核磁数据为:1h nmr(500mhz,cd4o)δ3.30~3.25(m,2h),3.01(dd,j=7.1,1.2hz,2h),2.97~2.92(m,3h),2.89(d,j=7.2hz,1h),2.87~2.82(m,2h),0.77(t,j=1.0hz,2h),-1.41(s,6h),-1.71(s,3h),-2.00(s,3h)。ms(esi):m/z 464[m h]
;
[0131]
s8:化合物9的制备,反应式参见下式14:
[0132][0133]
室温下,反应瓶中依次投入乙醇(200ml)、化合物8(10.00g,21.57mmol)、10%pd/c(2.00g)和甲酸铵(6.82g,107.85mmol),开启加热和搅拌。升温至回流,保温反应30min。保温结束后,tlc监控反应进程至无原料残留,停止反应。趁热过滤,滤饼用预热的异丙醇(20ml
×
3)洗涤至无产品。滤液减压浓缩至干。浓缩物用10%meoh/dcm(50ml)溶解,饱和盐水洗涤,无水硫酸钠干燥,减压浓缩至干得到7.88g白色固体(化合物9),收率97.82%,其核磁数据为:1h nmr(500mhz,cdcl3)δ7.73(d,j=8.8hz,2h),7.67(d,j=2.2hz,1h),7.30~7.25(m,3h),3.03(s,6h),2.73(s,3h),2.43(s,3h)。ms(esi):m/z 374[m h]
。
[0134]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
