1.本发明涉及技术领域,更具体地,涉及一种染色体臂端粒长度估计方法及系统。
背景技术:
2.端粒是位于真核生物染色体末端的含有多个鸟嘌呤(ttaggg)的串联重复序列,在绝大多数正常的人类细胞中,端粒会随着每次细胞分裂发生缩短,因而端粒被提出是限制细胞可以复制次数的一种标记物。端粒不是线性结构,由于许多关键蛋白质,端粒dna保持在环状结构中,这种结构用于保护染色体的末端。在肿瘤样品中利用全基因组测序数据估计端粒长度,发现端粒长度和肿瘤很多重要表型相关。
3.检测端粒长度可以帮助识别那些罹患与短端粒相关的疾病风险增加的人群,并且,通过有效的方法测定端粒的长度,对于衰老预警和癌症具有潜在的中大意义和价值。端粒在细胞分裂的每个循环中都会缩短,各种测量端粒长度的实验和计算方法被开发出来,实验方法主要包括端粒末端限制性片段分析trf(terminal restr-iction fragment,端粒末端限制性片段分析),定量pcr(quantitative pcr,qpcr),定量荧光原位杂交(quantitative fluoresc-ence in situ hybridization,q-fish)和单链端粒长度分析(single telomere length analysis,stela)。然而,trf的局限在于需要的dna量多,另外由于短端粒与探针结合的效率不高,trf对检测较短的端粒是不敏感的。qpcr的局限是需要扩增对照基因,而扩增的对照基因在基因组中是独特的,其拷贝数的变异和染色体的复制会改变基因拷贝数,从而显著改变t/s值。因此,qpcr法只适合用于二倍体和核型稳定的细胞和样本,转化细胞系和肿瘤组织样本是不适合的。而分裂间期q-fish不能分析每个染色体端粒长度和无端粒染色体,得出的也是平均端粒长度。stela方法需要具备单分子pcr技术经验,对技术操作要求较高,操作过程比较耗时法,不适合临床应用。比较分析这些端粒长度检测方法可以看出目前缺少一种单一的方法可以精确、简单、快速的检测端粒长度,因此端粒长度检测方法的选择需要根据具体的科学问题。
技术实现要素:
4.本发明为克服上述现有技术缺少可以精确、简单、快速的检测端粒长度的方法,提供一种染色体臂端粒长度估计方法及系统。
5.为解决上述技术问题,本发明的技术方案如下:
6.一种染色体臂端粒长度估计方法(telodiff),包括以下步骤:
7.获取10x基因组测序数据;计算所述10x基因组测序数据中整个10xg分子之间的长度差异不重叠的端粒数,或计算10xg分子重叠的映射dna片段的端粒数,将所述端粒数概率成正比到其当前估计的染色体臂级端粒长度分数charmtl,并重复上述计算直至染色体臂端粒长度分数charmtl收敛,输出染色体臂端粒长度分数charmtl。
8.作为优选方案,计算所述染色体臂端粒长度tl的分数的步骤包括:
9.计算所述10x基因组测序数据中10xg分子的平均长度la;
10.对于每个染色体臂,计算该染色体臂端粒邻近部分重叠的10xg分子中,其重叠部分的平均长度lm;
11.以所述平均长度la与所述平均长度lm的差值作为当前估计的染色体臂级端粒长度分数charmtl。
12.作为优选方案,所述方法还包括以下步骤:
13.更新当前估计的染色体臂端粒长度tl作为每个染色体臂分配的总端粒读数;重复上述计算直至所述染色体臂端粒长度tl的分数charmtl收敛,输出染色体臂端粒长度估计结果。
14.作为优选方案,所述方法还包括以下步骤:
15.构建随机森林模型,其中,以所述分数charmtl作为随机森林模型的预测标签,以包含全基因组测序数据中重复读数的频率x作为模型输入;
16.将全基因组测序端粒的读取频率,乘以10x genomics样品的总端粒读数和每个全基因组测序样品的总端粒读数的平均值的比率,然后将缩放的全基因组测序端粒频率进一步分位数归一化并转换为z分数后输入所述随机森林模型中进行训练,得到用于预测分数charmtl的随机森林模型。
17.进一步地,本发明还提出一种染色体臂端粒长度估计方法(teloem),包括以下步骤:
18.获取10x基因组测序数据;
19.将每个未映射的10xg分子分配到根据10x条码选择的具有可映射的染色体臂上;
20.读取所有完成分配的10xg分子相应的10x条码,从具有相同10x条码的10xg分子中读取次reads,对reads求和后将其概率成正比到当前估计的染色体臂级端粒长度分数charmtl;重复上述计算直至染色体臂端粒长度分数charmtl收敛,得到相应的染色体臂端粒长度tl。
21.作为优选方案,所述将每个未映射的10xg分子分配到根据10x条码选择的具有可映射的染色体臂上的步骤包括:
22.计算分配到任一染色体臂的概率,将所述概率大于预设阈值时相应的染色体臂作为具有可映射的染色体臂;
23.其中,分配到任一染色体臂的概率包括染色体臂的估计端粒长度占相同barcode选择的染色体臂估计端粒长度的和。
24.作为优选方案,所述方法还包括以下步骤:更新当前估计的染色体臂端粒长度tl作为每个染色体臂分配的总端粒读数;重复上述计算直至所述染色体臂端粒长度tl的分数charmtl收敛,得到相应的染色体臂端粒长度tl。
25.作为优选方案,所述方法还包括以下步骤:
26.构建随机森林模型,其中,以所述分数charmtl作为随机森林模型的预测标签,以包含全基因组测序数据中重复读数的频率x作为模型输入;
27.将全基因组测序端粒的读取频率,乘以10x genomics样品的总端粒读数和每个全基因组测序样品的总端粒读数的平均值的比率,然后将缩放的全基因组测序端粒频率进一步分位数归一化并转换为z分数后输入所述随机森林模型中进行训练,得到用于预测分数charmtl的随机森林模型。
28.进一步地,本发明还提出一种染色体臂端粒长度估计系统,包括存储器和处理器,所述存储器存储有计算机程序,所述处理器执行所述计算机程序时实现上述任一技术方案提出的染色体臂端粒长度估计方法的步骤。
29.与现有技术相比,本发明技术方案的有益效果是:本发明利用高通量数据估计单染色体臂的端粒长度,可以估计10x基因组测序数据以及高通量全基因组测序样品中的染色体臂水平的端粒长度,实现精确、简单、快速的端粒长度检测,适用于大型项目中染色体臂级端粒的研究。
附图说明
30.图1为实施例1的染色体臂端粒长度估计方法的流程图。
31.图2为实施例1的染色体臂端粒长度估计方法的原理图。
32.图3为实施例2的染色体臂端粒长度估计方法的流程图。
33.图4为实施例2的染色体臂端粒长度估计方法的原理图。
34.图5为实施例3中不同种族间染色体长度的差异数据图。
35.图6为实施例3中肿瘤和正常组织charmtls差异数据图。
具体实施方式
36.附图仅用于示例性说明,不能理解为对本专利的限制;
37.为了更好说明本实施例,附图某些部件会有省略、放大或缩小,并不代表实际产品的尺寸;
38.对于本领域技术人员来说,附图中某些公知结构及其说明可能省略是可以理解的。
39.下面结合附图和实施例对本发明的技术方案做进一步的说明。
40.实施例1
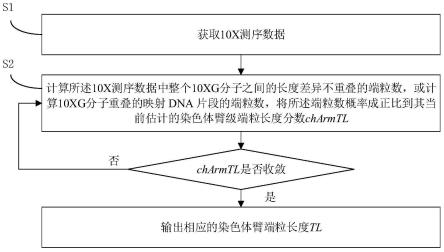
41.本实施例提出一种染色体臂端粒长度估计方法(telodiff),如图1所示,为本实施例的染色体臂端粒长度估计方法的流程图。
42.本实施例提出的染色体臂端粒长度估计方法中,包括以下步骤:
43.s1、获取10x基因组样品的测序数据。
44.s2、计算所述10x基因组测序数据中整个10xg分子之间的长度差异不重叠的端粒数,或计算10xg分子重叠的映射dna片段的端粒数,将所述端粒数概率成正比到其当前估计的染色体臂级端粒长度分数charmtl;
45.重复上述计算直至染色体臂端粒长度分数charmtl收敛,得到相应的染色体臂端粒长度tl。
46.本实施例中提出的上述染色体臂端粒长度估计方法telodiff,其中以10x基因组测序数据作为主体,是考虑到10x基因组测序通量高且linked reads可有效提供跨度超过几十kb甚至更长的信息,通过barcode overlap关系能够准确的区分单体型变异中不相邻的区域。理论上可以利用10x基因组测序估计每条染色体臂的端粒长度。
47.本实施例考虑端粒长度近似于整个10xg分子之间的长度差异不重叠的端粒数量组成的长度,或10xg分子重叠的映射dna片段的端粒数量组成的长度,将相应的端粒数概率
成正比到其当前估计的染色体臂级端粒长度分数charmtl,实现染色体臂端粒长度估计。
48.如图2所示,为本实施例的染色体臂端粒长度估计原理图。
49.其中,对于每个臂,计算所述10x基因组测序数据中10xg分子的平均长度la。对于每个染色体臂,计算该染色体臂端粒邻近部分重叠的10xg分子中,其重叠部分的平均长度lm。
50.以所述平均长度la与所述平均长度lm的差值作为当前估计的染色体臂级端粒长度分数charmtl。其伪代码如下所示:
[0051][0052]
进一步地,在一可选实施例中,所述方法还包括以下步骤:
[0053]
更新当前估计的染色体臂端粒长度tl作为每个染色体臂分配的总端粒读数;重复上述计算直至所述染色体臂端粒长度tl的分数charmtl收敛,得到相应的染色体臂端粒长度tl。
[0054]
进一步地,为了加速算法和为了降低10xg数据存储的成本,在一具体实施例中,仅使用1mb内映射到相应端粒的10x读取(类似结果使用从1mb到10mb的值,步长为1mb)来计算每个charmtl估计的背景10xg分子的大小。
[0055]
进一步地,在一可选实施例中,所述方法还包括以下步骤:
[0056]
s3.1、构建随机森林模型,其中,以所述分数charmtl作为随机森林模型的预测标签,以包含全基因组测序数据中重复读数的频率x作为模型输入。
[0057]
s3.2、将全基因组测序端粒的读取频率,乘以10x genomics样品的总端粒读数和每个全基因组测序样品的总端粒读数的平均值的比率,然后将缩放的全基因组测序端粒频率进一步分位数归一化并转换为z分数后输入所述随机森林模型中进行训练,得到用于预测分数charmtl的随机森林模型。
[0058]
具体地,本实施例以包含全基因组测序数据(ttaggg)n中重复读数的频率x作为随机森林模型的输入。在一具体实施例中,从52个细胞系样本中,以重复读数的频率x作为随机森林模型的输入,其中每个样本有92个变量。随机森林模型的输出为52个细胞系样本的charmtl。其伪代码如下所示:
[0059][0060]
本实施例中,利用高通量数据估计单染色体臂的端粒长度,可应用于大型项目中研究染色体臂级(charm,chromosome arm-level)端粒长度估计。
[0061]
实施例2
[0062]
本实施例提出一种染色体臂端粒长度估计方法(teloem),如图3所示,为本实施例的染色体臂端粒长度估计方法的流程图。
[0063]
本实施例提出的染色体臂端粒长度估计方法中,包括以下步骤:
[0064]
s1、获取10x基因组测序数据。
[0065]
s2、将每个未映射的10xg分子分配到根据10x条码选择的具有可映射的染色体臂上。
[0066]
s3、读取所有完成分配的10xg分子相应的10x条码,从具有相同10x条码的10xg分子中读取次reads,对reads求和后将其概率成正比到当前估计的染色体臂级端粒长度分数charmtl;重复上述计算直至染色体臂端粒长度分数charmtl收敛,得到相应的染色体臂端粒长度tl。
[0067]
本实施例提出的染色体臂端粒长度估计方法teloem中,将每个10x条码(bx)中未映射的端粒分配给具有相同条形码bx的分子中可映射的染色体臂,然后读取所有完成分配的10xg分子相应的10x条码,从具有相同10x条码的10xg分子中读取次reads,用每个染色体臂分配到的端粒部分10x分子的reads的和作为新的长度估计值。进一步,可选地采用期望最大化(em)迭代策略进行更新,得到相应的染色体臂端粒长度tl。
[0068]
如图4所示,为本实施例的染色体臂端粒长度估计方法的原理图。
[0069]
在一可选实施例中,所述将每个未映射的10xg分子分配到根据10x条码选择的具有可映射的染色体臂上的步骤包括:
[0070]
计算分配到任一染色体臂的概率,将所述概率大于预设阈值时相应的染色体臂作为具有可映射的染色体臂。其中,分配到任一染色体臂的概率包括染色体臂的估计端粒长度占相同barcode选择的染色体臂估计端粒长度的和。
[0071]
进一步地,在一可选实施例中,所述方法还包括以下步骤:
[0072]
更新当前估计的染色体臂端粒长度tl作为每个染色体臂分配的总端粒读数;重复上述计算直至所述染色体臂端粒长度tl的分数charmtl收敛,得到相应的染色体臂端粒长度tl。
[0073]
本实施例采用期望最大化(em)迭代策略构建得到,其中,所有charmtl都被初始化为相等。在每次迭代期间,将每个10x条码(bx)中未映射的端粒分配给具有相同条形码bx的分子中可映射的染色体臂,且概率与当前估计的charmtl除以charmtl的总和成正比。然后,将每个charmtl更新为每个臂的总分配端粒读数。重复这两个步骤直到所有charmtl收敛,得到相应的染色体臂端粒长度tl。其伪代码如下所示:
[0074]
[0075][0076]
进一步地,在一可选实施例中,所述方法还包括以下步骤:
[0077]
构建随机森林模型,其中,以所述分数charmtl作为随机森林模型的预测标签,以包含全基因组测序数据中重复读数的频率x作为模型输入。
[0078]
将全基因组测序端粒的读取频率,乘以10x genomics样品的总端粒读数和每个全基因组测序样品的总端粒读数的平均值的比率,然后将缩放的全基因组测序端粒频率进一步分位数归一化并转换为z分数后输入所述随机森林模型中进行训练,得到用于预测分数charmtl的随机森林模型。
[0079]
实施例3
[0080]
本实施例将实施例1、2提出的染色体臂端粒长度估计方法telodiff、teloem应用于人群样品。
[0081]
最近的一项研究发现,非裔美国人的平均端粒长度值比欧洲裔美国人要长。从10个xg样本中估计的charmtls,我们观察到欧洲人(eur)的charmtls总共比非洲人(afr)的要长。此外,eur、278名东亚人(eas)和ad混合美国人(amr)表现出相似的charmtls,而afr和南亚人(sas)表现出相似的charmtls。然后,我们比较了每组的eur和afr,尽管eur倾向于显示大多数染色体臂的比afr长,但只有7个染色体臂的一般显著(p值《0.05和》0.01),一些染色体臂的afr往往比eur长。因此,我们推测,扩大种群样本将进一步提供深入了解染色体长度的种群差异。
[0082]
本实施例telodiff、teloem应用于illumina的项目选择的1000个gp样本(n=150)的charmtl估计。如图5所示,为本实施例的不同种族间染色体长度的差异数据图。实验数据显示,非洲人287(afr)中15个charmtls比欧洲人(eur)更长,23个charmtls在afr中更短。此外,在afr 28中,三个染色体臂(17p、19p和8p)倾向于更长(但不显著)。
[0083]
本实施例同样将telodiff、teloem应用于大规模癌症样本中,检测了平均端粒长度。为了研究癌症中的charmtls,我们还试图预测来自癌症基因组图谱肝细胞癌(tcga-lihc)项目的肝癌wgs样本(54个肿瘤和54个匹配的正常样本)中的charmtls。
[0084]
如图6所示,为本实施例的肿瘤和正常组织charmtls差异数据图。实验数据显示,肿瘤中的charmtl更短(19个短臂和7个长臂),这与之前的报道一致,即肝脏肿瘤显示的平均tls比正常组织短40。预测的charmtl与肿瘤纯度呈负相关。
[0085]
由此可见,本发明提出的染色体臂端粒长度估计方法能够有效,且精确、简单、快速的检测端粒长度。
[0086]
实施例4
[0087]
本实施例提出一种染色体臂端粒长度估计系统,其中包括存储器和处理器,所述存储器存储有计算机程序,所述处理器执行所述计算机程序时实现实施例1提出的染色体臂端粒长度估计方法的步骤。
[0088]
具体地,所述处理器执行所述计算机程序时,执行以下步骤:
[0089]
s1、获取10x基因组测序数据。
[0090]
s2、计算所述10x基因组测序数据中整个10xg分子之间的长度差异不重叠的端粒数,或计算10xg分子重叠的映射dna片段的端粒数,将所述端粒数概率成正比到其当前估计的染色体臂级端粒长度分数charmtl;
[0091]
重复上述计算直至染色体臂端粒长度分数charmtl收敛,得到相应的染色体臂端粒长度tl。
[0092]
其中,对于每个臂,计算所述10x基因组测序数据中10xg分子的平均长度la。对于每个染色体臂,计算该染色体臂端粒邻近部分重叠的10xg分子中,其重叠部分的平均长度lm。
[0093]
以所述平均长度la与所述平均长度lm的差值作为当前估计的染色体臂级端粒长度分数charmtl。
[0094]
进一步地,更新当前估计的染色体臂端粒长度tl作为每个染色体臂分配的总端粒读数;重复上述计算直至所述染色体臂端粒长度tl的分数charmtl收敛,得到相应的染色体
臂端粒长度tl。
[0095]
实施例5
[0096]
本实施例提出一种染色体臂端粒长度估计系统,其中包括存储器和处理器,所述存储器存储有计算机程序,所述处理器执行所述计算机程序时实现实施例2提出的染色体臂端粒长度估计方法的步骤。
[0097]
具体地,所述处理器执行所述计算机程序时,执行以下步骤:
[0098]
s1、获取10x基因组测序数据。
[0099]
s2、将每个未映射的10xg分子分配到根据10x条码选择的具有可映射的染色体臂上。
[0100]
s3、读取所有完成分配的10xg分子相应的10x条码,从具有相同10x条码的10xg分子中读取次reads,对reads求和后将其概率成正比到当前估计的染色体臂级端粒长度分数charmtl;重复上述计算直至染色体臂端粒长度分数charmtl收敛,得到相应的染色体臂端粒长度tl。
[0101]
其中,所述将每个未映射的10xg分子分配到根据10x条码选择的具有可映射的染色体臂上的步骤包括:
[0102]
计算分配到任一染色体臂的概率,将所述概率大于预设阈值时相应的染色体臂作为具有可映射的染色体臂。其中,分配到任一染色体臂的概率包括染色体臂的估计端粒长度占相同barcode选择的染色体臂估计端粒长度的和。
[0103]
进一步地,更新当前估计的染色体臂端粒长度tl作为每个染色体臂分配的总端粒读数;重复上述计算直至所述染色体臂端粒长度tl的分数charmtl收敛,得到相应的染色体臂端粒长度tl。
[0104]
相同或相似的标号对应相同或相似的部件;
[0105]
附图中描述位置关系的用语仅用于示例性说明,不能理解为对本专利的限制;
[0106]
显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。