1.本发明属于半导体技术领域,涉及一种半导体硅前驱体的反应装置及方法,尤其涉及一种利用副产物再生三甲硅烷基胺的反应装置及方法。
背景技术:
2.三甲硅烷基胺(trisilylamine,tsa)的结构式为(sih3)3n,是一种易挥发、无色、可自燃以及易水解的液体。三甲硅烷基胺是半导体制造过程中重要的硅前驱体,可在没有直接等离子体激发的情况下进行膜生长,为半导体制造提供硅源和氮源,广泛用于高深宽比(har)的硅介电膜结构。
3.三甲硅烷基胺的合成方法主要包括:(1)单卤硅烷与氨反应制备tsa,具体分为液相法和气相法两种,各有优缺点;(2)热解全氢聚硅氮烷制备tsa;(3)通过氨基转移制备tsa。目前,工业上主要采用氯硅烷与氨反应来制备tsa的工艺路线,但在生产tsa的过程中,常伴有相当比例的副产物二甲硅烷基胺(disilylamine, dsa)生成,若dsa得不到有效利用,则会被当做危废处理。但目前还没有完备的利用副产物dsa再生制备tsa的方法。
4.氯硅烷与氨反应合成tsa的机理一般认为包括如下步骤:sih3cl 2nh3ꢀ→ꢀ
sih3nh
2 nh4clsih3cl sih3nh
2 nh3ꢀ→ꢀ
(sih3)2nh nh4clsih3cl (sih3)2nh nh3ꢀ→ꢀ
(sih3)3n nh4cl上述步骤中,dsa首先与氯硅烷反应得到(sih3)3nhcl中间体加合物,随后与另一氨分子反应生成tsa。
5.此外,也有研究认为,氯硅烷与氨合成tsa的反应机理按照以下方式进行:2sih3nh2ꢀ→ꢀ
(sih3)2nh nh33(sih3)2nh
ꢀ→ꢀ
2(sih3)3n nh3上述反应机理均能够表明,dsa能够向tsa进行自发转化,但dsa向tsa的自发转发需要一定的条件。例如,dsa在气相中不会转化为tsa,在液相中这种转化会缓慢地发生。有报道在0℃液相状态下,dsa在72小时有超过80%的比例转化成tsa。但在液态dsa自发转化tsa的过程中,会伴有nh3的生成,生成的nh3会降低tsa的稳定性:nnh
3 3(sih3)3n
ꢀ→ꢀ
3sih
4 (sih3nsih2)
3 nnh3(sih3nsih2)
3 xnh3ꢀ→ꢀ
ysih
4 znh
3 [polymeric materials]此外,虽然dsa与氨气在气相下不发生反应,但在液相条件下,dsa就会被氨全部破坏,分解成硅烷和少量氨。
[0006]
随tsa生产过程而伴生的副产物dsa,如不能得到有效利用,将会被当作危险废物处理而浪费掉。因此,为有效利用副产物二甲硅烷基胺,需要开发一种利用副产物dsa,安全、稳定、可靠地再生三甲硅烷基胺的反应装置和方法。
技术实现要素:
[0007]
本发明的目的在于提供一种利用副产物再生三甲硅烷基胺的反应装置及方法,该反应装置和方法能够实现对tsa生产过程中副产物dsa的充分利用,避免了dsa作为危废的浪费,符合绿色化工要求,降低了环境风险。
[0008]
为达到此发明目的,本发明采用以下技术方案:第一方面,本发明提供了一种利用副产物再生三甲硅烷基胺的方法,所述方法包括如下步骤:(1)均匀混合二甲硅烷基胺、氯硅烷与溶剂,然后匀速通入氨气;氨气通入结束后,反应至氯硅烷消耗完毕,得到反应液;(2)减压蒸馏步骤(1)所得反应液,氨气收集在冷阱中,轻相储罐中得到蒸馏液;(3)步骤(2)所得蒸馏液依次经过滤、第一精馏与第二精馏,得到所述三甲硅烷基胺。
[0009]
本发明提供的方法,以生产tsa过程中的副产物dsa为原料,配合对tsa生产过程中条件的调控,经氯硅烷化、氨置换及分离纯化,能够安全、稳定以及可靠的生产tsa,避免了dsa作为危废的浪费,从而使该反应装置生产tsa的过程符合绿色化工的要求,降低了环境风险,具有工业应用价值。
[0010]
另外,本发明提供的方法在减压蒸馏过程中蒸出多余的氨,则减少了后续步骤进行过程中对tsa产品造成的破坏。
[0011]
优选地,步骤(1)所述二甲硅烷基胺与氯硅烷的质量比为1:(0.9-1.8),例如可以是1:0.9、1:1、1:1.2、1:1.5、1:1.6或1:1.8,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0012]
优选地,步骤(1)所述二甲硅烷基胺与溶剂的质量比为1:(3-10),例如可以是1:3、1:4、1:5、1:6、1:7、1:8、1:9或1:10,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。反应中溶剂加入过少,不利于分散原料;加入过多,影响dsa转化tsa的生产效率。因此步骤(1)所述二甲硅烷基胺与溶剂的质量比优选为1:(3-10)。
[0013]
优选地,步骤(1)所述二甲硅烷基胺与氨气的质量比为1:(0.25-0.6),例如可以是1:0.25、1:0.3、1:0.4、1:0.5或1:0.6,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0014]
优选地,步骤(1)所述均匀混合的温度为-50℃至0℃,例如可以是-50℃、-40℃、-30℃、-20℃、-10℃或0℃,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0015]
氯硅烷在常温常压下是一种具有刺激性恶臭的无色气体,化学性质非常活泼,能够与水、醇、酚、氨、硅醇以及有机酸等含活泼氢的化合物发生剧烈的放热反应,遇明火或高热时,甚至会发生燃烧或爆炸。
[0016]
在三甲硅烷基胺生产过程中,传质传热受到限制,单纯的混合、制冷等方式很难及时分散剧烈反应产生的热量,而反应温度较高,且存在氨或氯化铵的条件下,生成聚硅氮烷的副反应更容易发生。当步骤(1)所述均匀混合的温度高于0℃时,非常容易导致反应温度过高,引起生成的聚硅氮烷比例增多。当均匀混合的温度过低时,氯硅烷与氨反应生成tsa的速率变慢,影响tsa的转化效率;而且,较低的混合温度也使溶剂的选择较为困难,不利于
分散的顺利进行。因此,为了安全稳定地生产三甲硅烷基胺,本技术步骤(1)所述均匀混合的温度优选为-50℃至0℃。
[0017]
dsa的沸点是36℃,在-23℃的蒸气压是94.5mmhg,在-44.5℃的蒸气压是28.9mmhg。因此优选反应在低温下进行,尤其在0℃以下,确保dsa组分分散在中溶剂中;低于-50℃的反应温度将不利选择合适的溶剂分散。因此,本技术步骤(1)所述均匀混合的温度优选为-50℃至0℃。
[0018]
优选地,步骤(1)所述均匀混合的方法包括搅拌,所述搅拌的转速为50-300r/min,例如可以是50r/min、100r/min、150r/min、200r/min、250r/min或300r/min,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0019]
优选地,步骤(1)所述通入氨气的过程中伴随搅拌,搅拌的转速为50-300r/min,例如可以是50r/min、100r/min、150r/min、200r/min、250r/min或300r/min,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0020]
优选地,步骤(1)所述反应过程中伴随搅拌,搅拌的转速为50-300r/min,例如可以是50r/min、100r/min、150r/min、200r/min、250r/min或300r/min,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0021]
优选地,步骤(1)所述溶剂的沸点在100℃以上,凝固点在-25℃以下。
[0022]
优选地,步骤(1)所述溶剂包括正辛烷、正壬烷、正癸烷、甲苯、二甲苯或d系列溶剂油中的任意一种或至少两种的组合,典型但非限制性的组合包括正辛烷与正壬烷的组合,正壬烷与甲苯的组合,甲苯与二甲苯的组合,二甲苯与d系列溶剂油的组合,正辛烷、正壬烷与甲苯的组合,甲苯、二甲苯与d系列溶剂油的组合,或,正辛烷、正壬烷、甲苯、二甲苯或d系列溶剂油的组合,优选为正壬烷。
[0023]
本发明选择的溶剂在减压蒸馏完全蒸出产物时,不会蒸出过多的溶剂,避免反应装置内因溶剂量的减少而影响副产物氯化铵的分散,还能够保证精馏效率。其中,正辛烷的沸点为125℃,凝固点为-57℃;正癸烷的沸点为174℃,凝固点为-30℃;甲苯的沸点为110.6℃,凝固点为-94.9℃;二甲苯的沸程为135-145℃,邻二甲苯的凝固点为-25.2℃,间二甲苯的凝固点为-48℃,对二甲苯的凝固点为12-13℃;以d40为例,其沸程为150-210℃,冰点为-40℃。
[0024]
而正壬烷的沸点为151℃,且凝固点为-53℃,其在步骤(1)均匀混合的过程中不凝固结块,可稳定分散反应。而且,正壬烷在0℃的蒸气压为0.1kpa,当减压蒸馏完全蒸出产物时,减少了溶剂的蒸出量;在第二精馏时,正壬烷与tsa的沸点以及蒸气压差别较大,还能够保证精馏效率。因此,本发明的溶剂优选为正壬烷。
[0025]
优选地,步骤(1)所述通入氨气的时间为3-12h,例如可以是3h、5h、6h、8h、10h或12h,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。当通入氨气的时间低于3h时,体系内的氯硅烷与氨的反应过快,反应产生的热量不利于及时分散,体系温度容易冲高,进而导致生成的tsa容易遭破坏,产生更多的聚硅氮烷;通入氨气的时间超过12h时,则会影响dsa转化tsa的生产效率。
[0026]
此外,虽然dsa与氯硅烷在初始物料混合阶段很快反应,但再生三甲硅烷基胺的反应过程中也存在dsa组分的生成;如通入氨过快,体系内过量的氨将会破坏dsa的稳定性,导致分解产生硅烷;如通入氨过慢,又会影响dsa转化tsa的生产效率。因此,优选步骤(1)所述
通入氨气的时间为3-12h。
[0027]
优选地,步骤(1)所述反应的绝对压力为1-6bar,例如可以是1bar、2bar、3bar、4bar、5bar或6bar,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0028]
本发明所述反应的绝对压力是指反应过程中反应装置内的压力,由于氨气的消耗,反应的绝对压力存在波动,因此,本发明所述反应的绝对压力为反应过程中的平衡压力。当通入氨气无法维持反应的绝对压力时,通入氮气和/或惰性气体进行补偿。
[0029]
本发明步骤(1)中匀速通入氨气的温度以及反应时的温度均与均匀混合的温度相同。本发明提供的制备方法通过对反应条件的控制,避免了危险原料氯硅烷的回收步骤,也避免了氯硅烷与dsa反应造成的铵盐沉淀问题;而且,反应过程在-50℃至0℃的条件下进行,避免了反应无法及时散热造成的危险,也减少了聚硅氮烷的生成。
[0030]
优选地,步骤(2)所述减压蒸馏的绝对压力为0.2-0.8bar,例如可以是0.2bar、0.3bar、0.4bar、0.5bar、0.6bar、0.7bar或0.8bar,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0031]
优选地,步骤(2)所述减压蒸馏的温度为-30℃至20℃,例如可以是-30℃、-20℃、-10℃、0℃、10℃或20℃,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0032]
本发明所述减压蒸馏的温度为反应装置的温度;作为优选的技术方案,为了控制溶剂的蒸出量,与反应装置配套的冷凝装置的温度为-20℃至0℃,例如可以是-20℃、-15℃、-10℃、-5℃或0℃,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0033]
本发明步骤(2)所述减压蒸馏的终点为使dsa以及tsa完全蒸出。
[0034]
本发明所述减压蒸馏时,需要在dsa与tsa蒸出体系之前(dsa沸点36℃,tsa沸点52℃)蒸出多余的氨,因此需要减压蒸馏在较低温度下开始;蒸氨过程为吸热过程,蒸氨进行过程中,反应装置内的温度会进一步降低,因此,若减压蒸馏的温度低于-30℃时,随着减压蒸馏的进行,体系的温度会逐渐降低至氨的液化点(-33.5℃)以下,影响氨的进一步蒸出;当温度低于溶剂的凝固点时,会导致体系粘稠甚至溶剂结块,影响产物的蒸馏;而当减压蒸馏的温度超过20℃时,则会使氨与副产物氯化铵加速tsa的分解,增加聚硅氮烷的生成。因此,需要控制减压蒸馏的温度为-30℃至20℃。
[0035]
本发明控制减压蒸馏的条件,不仅能够实现产品的减压蒸馏,还能够蒸出多余的氨,减少纯化过程中对产品tsa造成的破坏。
[0036]
优选地,步骤(3)所述过滤为增压过滤,增压过滤的气体压强为1-5bar,例如可以是1bar、2bar、3bar、4bar或5bar,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0037]
本发明制备方法的产物dsa与tsa的饱和蒸气压较高,且氯化铵对tsa与dsa还具有捕集作用,采用常规的减压过滤或常压过滤,均无法实现对dsa以及tsa的充分回收;而增压过滤还能够缩短过滤时间,避免了产物被催化分解的风险。
[0038]
本发明所述第一精馏在第一精馏装置中进行,通过第一精馏,使第一精馏装置的重组分中不含dsa;本发明所述第二精馏在第二精馏装置中进行,通过第二精馏,使第二精馏装置的塔顶得到产品tsa。
[0039]
优选地,步骤(3)所述第一精馏的塔釜温度为90-100℃,塔顶温度为45-50℃。
[0040]
本发明所述第一精馏的塔釜温度为90-100℃,例如可以是90℃、92℃、95℃、96℃、
98℃或100℃,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0041]
本发明所述第一精馏的塔顶温度为45-50℃,例如可以是45℃、46℃、47℃、48℃、49℃或50℃,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0042]
本发明不对第一精馏的回流比进行具体限定,只要能够使第一精馏装置的重组分中不含dsa,而且是第一精馏的塔釜温度以及塔顶温度符合要求即可。
[0043]
优选地,步骤(3)所述第二精馏的塔釜温度为110-120℃,塔顶温度为55-60℃。
[0044]
本发明所述第二精馏的塔釜温度为110-120℃,例如可以是110℃、112℃、115℃、116℃、118℃或120℃,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0045]
本发明所述第二精馏的塔顶温度为55-60℃,例如可以是55℃、56℃、57℃、58℃、59℃或60℃,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0046]
本发明不对第二精馏的回流比进行具体限定,只要能够实现第二精馏得到产品tsa即可。示例性的,所述第二精馏的的回流比为8-12,例如可以是8、9、10、11或12,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0047]
作为优选地技术方案,第一精馏得到的轻组分作为二甲硅烷基胺的供给源,与氯硅烷、溶剂进行反应。
[0048]
第二方面,本发明提供了一种反应装置,用于第一方面所述利用副产物再生三甲硅烷基胺的方法,所述反应装置包括反应单元、减压蒸馏单元、过滤单元、精馏单元以及清洁单元;所述反应单元包括反应装置、二甲硅烷基胺供给装置、溶剂供给装置、氨气进料管道以及氯硅烷进料管道;所述反应装置设置有搅拌装置;所述溶剂供给装置、氨气进料管道以及氯硅烷进料管道分别独立地与反应装置连接;所述氨气进料管道设置有联动阀门;所述联动阀门与搅拌装置连锁;所述减压蒸馏单元包括第一冷凝装置、第二冷凝装置、真空装置、冷阱以及轻相储罐;所述第一冷凝装置、第二冷凝装置、冷阱以及真空装置依次连接,且第一冷凝装置的进料口与反应装置的出气口连接;所述第二冷凝装置的冷凝液出口与轻相储罐连接;所述冷阱与轻相储罐的连接管路连接有平衡气管路;所述过滤单元包括第一过滤系统、第二过滤系统、产品滤液储罐以及溶剂回收储罐;第一过滤系统与第二过滤系统分别独立地包括至少2个并联的过滤装置,过滤装置的进口与出口分别连通气体管路;所述第一过滤系统连通轻相储罐与产品滤液储罐;所述产品滤液储罐与精馏单元连接;所述第二过滤系统连通反应装置与溶剂回收储罐;所述精馏单元包括至少2级串联连接的精馏系统;最后一级精馏系统中,精馏装置的底部出液口与溶剂回收储罐连接;所述清洁单元包括气体清洁管路;所述气体清洁管路分别与氨气进料管道、氯硅烷进料管道连接。
[0049]
本发明所述联动阀门与搅拌装置连锁是指,联动阀门的启闭控制氨气进料管道中的进气,当搅拌装置停止工作时,联动阀门关闭;当搅拌装置运行时,联动阀门根据氨气的进气需求开启或关闭。本发明通过联动阀门与搅拌装置的连锁,避免了继续通入氨气造成的氯硅烷与局部浓度过高的氨之间的剧烈反应,避免了散热不及时引发事故的风险。
[0050]
本发明所述反应装置为设置有控温部的反应装置,本发明不对控温部的结构做具
体限定,只要能够实现对反应装置的控温即可。示例性的,所述控温部包括但不限于换热盘管和/或换热夹层。
[0051]
本发明提供的第一过滤系统与第二过滤系统分别独立地包括至少2个并联的过滤装置,所述至少2个并联的过滤装置的设置能够保证过滤系统连续运行,避免其中任一过滤装置的维护对整个合成装置稳定运行的影响。
[0052]
其中第一过滤系统包括至少2个并联的过滤装置,例如可以是2个、3个、4个、5个或6个,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0053]
其中第二过滤系统包括至少2个并联的过滤装置,例如可以是2个、3个、4个、5个或6个,但不限于所列举的数值,数值范围内其它未列举的数值同样适用。
[0054]
本发明通过冷阱以及平衡气管路的设置,避免了反应装置内的真空度过低,从而避免了产物被吸入真空装置,保证了真空装置的稳定运转。
[0055]
本发明通过气体清洁管路与氨气进料管道连接,当氨气进料管道设置于反应装置的末端存在氯化铵附着、沉积时,能够实现关闭氨气后,采用保护性气体对氨气进料管道进行冲扫,避免了氯化铵的积聚。
[0056]
本发明所述气体清洁管路、平衡气管路以及气体管路中所用气体分别独立地包括氮气和/或惰性气体,所述惰性气体包括氦气、氖气或氩气中的任意一种或至少两种的组合,典型但非限制性的组合包括氦气与氖气的组合,氖气与氩气的组合,氦气与氩气的组合,或氦气、氖气与氩气的组合。
[0057]
示例性的,本发明所述溶剂供给装置为本领域常规的供给溶剂的装置,包括但不限于溶剂储罐以及配套的溶剂输送装置,本发明在此不做具体限定,只要能够实现反应装置中溶剂的供给即可。
[0058]
示例性的,本发明所述二甲硅烷基胺供给装置为本领域常规的供给二甲硅烷基胺的装置,包括但不限于二甲硅烷基胺储罐以及配套的输送装置,本发明在此不做具体限定,只要能够实现反应装置中二甲硅烷基胺的供给即可。
[0059]
应用本发明提供的合成装置时:(a)首先利用清洁单元吹扫合成装置,保证合成装置内干燥清洁;然后控制反应装置的温度至工艺温度,搅拌装置搅拌条件下,溶剂供给装置向反应装置内提供溶剂,二甲硅烷基胺供给装置向反应装置提供二甲硅烷基胺;利用氯硅烷进料管道向反应装置提供氯硅烷,氯硅烷进料结束后,清洁单元吹扫氯硅烷进料管道;(b)搅拌装置搅拌条件下,利用氨气进料管道均速向反应装置中供给氨气,至氨气供给量符合工艺要求,氨气进料结束后,清洁单元吹扫氨气进料管道;氨气供给结束后,搅拌装置持续搅拌,直至反应装置内的物料处理完毕;(c)反应装置缓慢泄压,然后利用真空装置与清洁装置供给的气体维持反应装置内的压力平衡,通过反应装置、第一冷凝装置以及第二冷凝装置的温度控制,使反应装置内的nh3、dsa、tsa、少量溶剂以及夹带的nh4cl转移出反应装置;nh3经过第一冷凝装置、第二冷凝装置的降温后,由冷阱收集;dsa、tsa、少量溶剂以及夹带的nh4cl经过第一冷凝装置与第二冷凝装置,由轻相储罐收集;(d)收集在轻相储罐中的产品混合物经过第一过滤系统的过滤处理,分离出固体nh4cl,剩余的产品滤液进入产品滤液储罐;产品滤液储罐中的产品滤液依次流经第一精馏
系统与第二精馏系统,调整第一精馏系统与第二精馏系统的参数条件,使第一精馏系统的重组分中不含dsa;第一精馏系统的重组分经过第二精馏系统处理,得到产品tsa以及收集于溶剂回收储罐的回收溶剂;(e)反应装置内的剩余溶剂中含有氯化铵,经过第二过滤系统的过滤处理,分离出固体氯化铵,过滤得到的溶剂回收于溶剂回收储罐。
[0060]
步骤(d)与步骤(e)不分先后顺序。
[0061]
为了副产物氯化铵能够良好分散,本发明所述合成装置的使用过程中,搅拌装置持续搅拌;若搅拌装置意外停止,通过联动阀门的使用,保证搅拌停止状态下,无氨气进入反应装置。
[0062]
优选地,本发明中的氨气进料管道的末端设置于反应装置的液面以下,且不影响搅拌分散。
[0063]
优选地,本发明所述氨气进料管道的材质应不易被副产物氯化铵盐附着以致阻塞管路,包括但不限于氟塑料。
[0064]
优选地,所述第一过滤系统包括并联连接的第一过滤装置与第二过滤装置。
[0065]
优选地,所述第二过滤系统包括并联连接的第三过滤装置与第四过滤装置。
[0066]
优选地,所述精馏单元包括串联连接的第一精馏系统与第二精馏系统;所述第一精馏系统包括第一精馏装置以及与第一精馏装置的轻组分出料口连接的第一产品储存装置;所述第一产品储存装置与二甲硅烷基胺供给装置之间设置连接管路;所述第二精馏系统包括第二精馏装置以及与第二精馏装置的轻组分出料口连接的第二产品储存装置。
[0067]
应用本发明所述精馏单元时,控制第一精馏装置的条件,使第一精馏装置内的重组分不含dsa,而主要为dsa的轻组分收集于第一产品储存装置;第一精馏装置中的重组分于第二精馏装置内进行处理,所得轻组分为产品tsa,其收集于第二产品储存装置中。
[0068]
本发明所述第一精馏装置包括但不限于第一精馏塔,所述第一产品储存装置包括但不限于第一产品储罐;本发明所述第二精馏装置包括但不限于第二精馏塔,所述第二产品储存装置包括但不限于第二产品储罐。
[0069]
优选地,所述溶剂供给装置与反应装置的连接管路还与溶剂回收储罐连接。
[0070]
本发明通过使溶剂供给装置与反应装置的连接管路与溶剂回收储罐连接,实现了溶剂回收储罐中溶剂的回收利用。
[0071]
优选地,所述联动阀门为电磁阀。
[0072]
优选地,所述轻相储罐设置有计量件。
[0073]
示例性的,本发明所述轻相储罐设置有计量件是指,轻相储罐设置有液位计、流量计或轻相储罐中带有刻度。通过计量件的设置,能够便于操作人员对减压蒸馏的蒸出量进行观察,避免蒸出过多溶剂对反应装置中nh4cl的分散及出料的不利影响,本领域技术人员能够根据实际需要,对计量件进行合理地选择。
[0074]
相对于现有技术,本发明具有以下有益效果:(1)本发明提供的方法以生产tsa过程中的副产物dsa为原料,配合对tsa生产过程中条件的调控,经氯硅烷化、氨置换及分离纯化,能够安全、稳定以及可靠的生产tsa,避免
了dsa作为危废的浪费,从而使该反应装置生产tsa的过程符合绿色化工的要求,降低了环境风险,具有工业应用价值;(2)本发明提供的利用副产物再生三甲硅烷基胺的反应装置,不仅能够实现dsa的充分利用,还能够稳定安全的实现tsa的生产,实现了对传统氯硅烷与氨制备tsa合成路线的改进,具有工业应用价值。
附图说明
[0075]
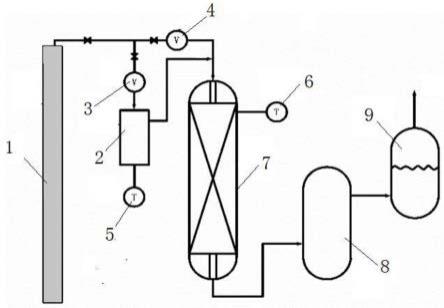
图1为本发明提供的利用副产物再生三甲硅烷基胺的反应装置的示意图。
[0076]
其中:11,反应装置;12,溶剂供给装置;13,二甲硅烷基胺供给装置;21,第一冷凝装置;22,第二冷凝装置;23,冷阱;24,真空装置;25,轻相储罐;31,第一过滤装置;32,第二过滤装置;33,产品滤液储罐;34,第三过滤装置;35,第四过滤装置;36,溶剂回收储罐;41,第一精馏装置;42,第一产品储存装置;43,第二精馏装置;44,第二产品储存装置。
具体实施方式
[0077]
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0078]
为了清楚说明本发明的技术方案,具体实施方式中用于副产物再生三甲硅烷基胺的反应装置的结构示意图如图1所示,包括反应单元、减压蒸馏单元、过滤单元、精馏单元以及清洁单元;所述反应单元包括反应装置11、溶剂供给装置12、二甲硅烷基胺供给装置13、氨气进料管道以及氯硅烷进料管道;所述反应装置11设置有搅拌装置;所述溶剂供给装置12、氨气进料管道以及氯硅烷进料管道分别独立地与反应装置11连接;所述氨气进料管道设置有联动阀门;所述联动阀门与搅拌装置连锁;所述联动阀门为电磁阀;氨气进料管道的末端设置于反应装置11的液面以下,且不影响搅拌分散;氨气进料管道的材质为氟塑料;所述减压蒸馏单元包括第一冷凝装置21、第二冷凝装置22、真空装置24、冷阱23以及轻相储罐25;所述第一冷凝装置21、第二冷凝装置22、冷阱23以及真空装置24依次连接,且第一冷凝装置21的进料口与反应装置11的出气口连接;所述第二冷凝装置22的冷凝液出口与轻相储罐25连接;所述冷阱23与真空装置24的连接管路连接有平衡气管路;所述轻相储罐25设置有液位计;所述过滤单元包括第一过滤系统、第二过滤系统、产品滤液储罐33以及溶剂回收储罐36;所述第一过滤系统包括并联连接的第一过滤装置31与第二过滤装置32,第二过滤系统包括并联连接的第三过滤装置34与第四过滤装置35,第一过滤装置31、第二过滤装置32、第三过滤装置34以及第四过滤装置35的进口与出口分别连通气体管路;所述第一过滤系统连通轻相储罐25与产品滤液储罐33;所述产品滤液储罐33与精馏单元连接;所述第二过滤系统连通反应装置11与溶剂回收储罐36;所述精馏单元包括串联连接的第一精馏系统与第二精馏系统;所述第一精馏系统包括第一精馏装置41以及与第一精馏装置41的轻组分出料口连接的第一产品储存装置42;所述第二精馏系统包括第二精馏装置43以及与第二精馏装置43的轻组分出料口连接的第二产品储存装置44;第二精馏装置43的底部出液口与溶剂回收储罐36连接;所述第一精馏
装置41为第一精馏塔,第二精馏装置43为第二精馏塔,第一精馏塔的塔釜与第二精馏塔的进料口连通;所述第一产品储存装置42与二甲硅烷基胺供给装置13之间设置连接管路;所述溶剂供给装置12与反应装置11的连接管路与溶剂回收储罐36连接。
[0079]
所述清洁单元包括气体清洁管路;所述气体清洁管路分别与氨气进料管道以及氯硅烷进料管道连接。
[0080]
所述气体清洁管路、平衡气管路以及气体管路中所用气体分别独立地为氮气。
[0081]
应用合成装置时:(a)首先利用清洁单元吹扫合成装置,保证合成装置内干燥清洁;然后控制反应装置11的温度至工艺温度,搅拌装置搅拌条件下,溶剂供给装置12向反应装置11内提供溶剂,二甲硅烷基胺供给装置13向反应装置11提供二甲硅烷基胺;利用氯硅烷进料管道向反应装置11提供氯硅烷,氯硅烷进料结束后,清洁单元吹扫氯硅烷进料管道;(b)搅拌装置搅拌条件下,利用氨气进料管道均速向反应装置11中供给氨气,至氨气供给量符合工艺要求,氨气进料结束后,清洁单元吹扫氨气进料管道;氨气供给结束后,搅拌装置持续搅拌,直至反应装置11内的物料处理完毕;(c)反应装置11缓慢泄压,然后利用真空装置24与清洁装置供给的气体维持反应装置11内的压力平衡,通过反应装置11、第一冷凝装置21以及第二冷凝装置22的温度控制,使反应装置11内的氨气、dsa、tsa、少量溶剂以及夹带的nh4cl转移出反应装置11;氨气经过第一冷凝装置21、第二冷凝装置22的降温后,由冷阱23收集;dsa、tsa、少量溶剂以及夹带的nh4cl经过第一冷凝装置21与第二冷凝装置22,由轻相储罐25收集;(d)收集在轻相储罐25中的产品混合物经过第一过滤系统的过滤处理,分离出固体nh4cl,剩余的产品滤液进入产品滤液储罐33;产品滤液储罐33中的产品滤液依次流经第一精馏装置41与第二精馏装置43,调整第一精馏装置41与第二精馏装置43的参数条件,使第一精馏装置41的重组分中不含dsa;第一精馏装置41的重组分经过第二精馏装置43处理,得到产品tsa以及回收于溶剂回收储罐36的回收溶剂;(e)反应装置11内的剩余溶剂中含有氯化铵,经过第二过滤系统的过滤处理,分离出固体氯化铵,过滤得到的溶剂回收于溶剂回收储罐36。
[0082]
步骤(d)与步骤(e)不分先后顺序。
[0083]
为了副产物氯化铵能够良好分散,本发明所述合成装置的使用过程中,搅拌装置持续搅拌;若搅拌装置意外停止,通过联动阀门的使用,保证搅拌停止状态下,无氨气进入反应装置。
[0084]
实施例1本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,所述方法包括如下步骤:(1)反应装置内均匀混合二甲硅烷基胺、氯硅烷与正壬烷,然后匀速通入氨气;通入氨气的时间为7h;氨气通入结束后,反应至氯硅烷消耗完毕,得到反应液;所述反应的绝对压力为3bar;所述二甲硅烷基胺与氯硅烷的质量比为1:1.4;所述二甲硅烷基胺与正壬烷的质量比为1:6;所述二甲硅烷基胺与氨气的质量比为1:0.42;步骤(1)进行过程中,反应装置内的温度维持-30℃;
(2)减压蒸馏步骤(1)所得反应液,氨气收集在冷阱中,轻相储罐中得到蒸馏液;减压蒸馏的绝对压力为0.4bar,在-30℃至20℃的范围内调节反应装置的温度,顺序地引导反应装置内的氨气、dsa和tsa、少许正壬烷及夹带的痕量nh4cl;减压蒸馏时,第一冷凝装置与第二冷凝装置的温度分别独立地为-10℃;(3)步骤(2)所得蒸馏液依次经过滤、第一精馏与第二精馏,得到所述三甲硅烷基胺;所述第一精馏的塔釜温度为100℃,塔顶温度为50℃,回流比为21,塔板数为60;第二精馏的塔釜温度为120℃,塔顶温度为60℃,回流比为12,塔板数为60;为了副产物氯化铵的良好分散,所述方法进行过程中,搅拌装置持续搅拌,搅拌转速为200r/min。
[0085]
步骤(3)所述第一精馏所得轻组分回流至二甲硅烷基胺供给装置以备用。
[0086]
实施例2本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,所述方法包括如下步骤:(1)反应装置内均匀混合二甲硅烷基胺、氯硅烷与正壬烷,然后匀速通入氨气;通入氨气的时间为10h;氨气通入结束后,反应至氯硅烷消耗完毕,得到反应液;所述反应的绝对压力为4bar;所述二甲硅烷基胺与氯硅烷的质量比为1:1.6;所述二甲硅烷基胺与正壬烷的质量比为1:8;所述二甲硅烷基胺与氨气的质量比为1:0.5;步骤(1)进行过程中,反应装置内的温度维持-30℃;(2)减压蒸馏步骤(1)所得反应液,氨气收集在冷阱中,轻相储罐中得到蒸馏液;减压蒸馏的绝对压力为0.6bar,在-30℃至20℃的范围内调节反应装置的温度,顺序地引导反应装置内的氨气、dsa和tsa、少许正壬烷及夹带的痕量nh4cl;减压蒸馏时,第一冷凝装置与第二冷凝装置的温度分别独立地为0℃;(3)步骤(2)所得蒸馏液依次经过滤、第一精馏与第二精馏,得到所述三甲硅烷基胺;所述第一精馏的塔釜温度为95℃,塔顶温度为48℃,回流比为20,塔板数为60;第二精馏的塔釜温度为115℃,塔顶温度为58℃,回流比为10,塔板数为60;为了副产物氯化铵的良好分散,所述方法进行过程中,搅拌装置持续搅拌,搅拌转速为100r/min。
[0087]
步骤(3)所述第一精馏所得轻组分回流至二甲硅烷基胺供给装置以备用。
[0088]
实施例3本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,所述方法包括如下步骤:(1)反应装置内均匀混合二甲硅烷基胺、氯硅烷与正壬烷,然后匀速通入氨气;通入氨气的时间为5h;氨气通入结束后,反应至氯硅烷消耗完毕,得到反应液;所述反应的绝对压力为2bar;所述二甲硅烷基胺与氯硅烷的质量比为1:1.2;所述二甲硅烷基胺与正壬烷的质量比为1:4;所述二甲硅烷基胺与氨气的质量比为1:0.35;步骤(1)进行过程中,反应装置内的温度维持-30℃;(2)减压蒸馏步骤(1)所得反应液,氨气收集在冷阱中,轻相储罐中得到蒸馏液;减压蒸馏的绝对压力为0.3bar,在-30℃至20℃的范围内调节反应装置的温度,顺序地引导反应装置内的氨气、dsa和tsa、少许正壬烷及夹带的痕量nh4cl;减压蒸馏时,第一冷凝装置与
第二冷凝装置的温度分别独立地为-20℃;(3)步骤(2)所得蒸馏液依次经过滤、第一精馏与第二精馏,得到所述三甲硅烷基胺;所述第一精馏的塔釜温度为90℃,塔顶温度为45℃,回流比为18,塔板数为60;第二精馏的塔釜温度为110℃,塔顶温度为55℃,回流比为8,塔板数为60;为了副产物氯化铵的良好分散,所述方法进行过程中,搅拌装置持续搅拌,搅拌转速为250r/min。
[0089]
步骤(3)所述第一精馏所得轻组分回流至二甲硅烷基胺供给装置以备用。
[0090]
实施例4本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,所述方法包括如下步骤:(1)反应装置内均匀混合二甲硅烷基胺、氯硅烷与正壬烷,然后匀速通入氨气;通入氨气的时间为12h;氨气通入结束后,反应至氯硅烷消耗完毕,得到反应液;所述反应的绝对压力为6bar;所述二甲硅烷基胺与氯硅烷的质量比为1:1.8;所述二甲硅烷基胺与正壬烷的质量比为1:10;所述二甲硅烷基胺与氨气的质量比为1:0.6;步骤(1)进行过程中,反应装置内的温度维持-30℃;(2)减压蒸馏步骤(1)所得反应液,氨气收集在冷阱中,轻相储罐中得到蒸馏液;减压蒸馏的绝对压力为0.8bar,在-30℃至20℃的范围内调节反应装置的温度,顺序地引导反应装置内的氨气、dsa和tsa、少许正壬烷及夹带的痕量nh4cl;减压蒸馏时,第一冷凝装置与第二冷凝装置的温度分别独立地为-10℃;(3)步骤(2)所得蒸馏液依次经过滤、第一精馏与第二精馏,得到所述三甲硅烷基胺;所述第一精馏的塔釜温度为100℃,塔顶温度为50℃,回流比为21,塔板数为60;第二精馏的塔釜温度为120℃,塔顶温度为60℃,回流比为12,塔板数为60;为了副产物氯化铵的良好分散,所述方法进行过程中,搅拌装置持续搅拌,搅拌转速为300r/min。
[0091]
步骤(3)所述第一精馏所得轻组分回流至二甲硅烷基胺供给装置以备用。
[0092]
实施例5本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,所述方法包括如下步骤:(1)反应装置内均匀混合二甲硅烷基胺、氯硅烷与正壬烷,然后匀速通入氨气;通入氨气的时间为3h;氨气通入结束后,反应至氯硅烷消耗完毕,得到反应液;所述反应的绝对压力为1bar;所述二甲硅烷基胺与氯硅烷的质量比为1:0.9;所述二甲硅烷基胺与正壬烷的质量比为1:3;所述二甲硅烷基胺与氨气的质量比为1:0.25;步骤(1)进行过程中,反应装置内的温度维持-30℃;(2)减压蒸馏步骤(1)所得反应液,氨气收集在冷阱中,轻相储罐中得到蒸馏液;减压蒸馏的绝对压力为0.2bar,在-30℃至20℃的范围内调节反应装置的温度,顺序地引导反应装置内的氨气、dsa和tsa、少许正壬烷及夹带的痕量nh4cl;减压蒸馏时,第一冷凝装置与第二冷凝装置的温度分别独立地为-10℃;(3)步骤(2)所得蒸馏液依次经过滤、第一精馏与第二精馏,得到所述三甲硅烷基胺;所述第一精馏的塔釜温度为100℃,塔顶温度为50℃,回流比为21,塔板数为60;第二精
馏的塔釜温度为120℃,塔顶温度为60℃,回流比为12,塔板数为60;为了副产物氯化铵的良好分散,所述方法进行过程中,搅拌装置持续搅拌,搅拌转速为50r/min。
[0093]
步骤(3)所述第一精馏所得轻组分回流至二甲硅烷基胺供给装置以备用。
[0094]
实施例6本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,除了步骤(1)中二甲硅烷基胺与正壬烷的质量比为1:0.7外,其余均与实施例1相同。
[0095]
本实施例中,由于溶剂比例过少,dsa与氯硅烷反应的中间产物不能分散,出现结块的问题,三甲硅烷基胺无法合成。
[0096]
实施例7本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,除了步骤(1)中二甲硅烷基胺与正壬烷的质量比为1:2外,其余均与实施例1相同。
[0097]
本实施例中,由于溶剂比例不足以分散dsa与氯硅烷反应的中间产物,出现结块,三甲硅烷基胺无法合成。
[0098]
实施例8本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,除步骤(1)进行过程中,反应装置内的温度维持-50℃外,其余均与实施例1相同。
[0099]
实施例9本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,除步骤(1)进行过程中,反应装置内的温度维持-10℃外,其余均与实施例1相同。
[0100]
实施例10本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,除步骤(1)进行过程中,反应装置内的温度维持-0℃外,其余均与实施例1相同。
[0101]
实施例11本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,除步骤(1)进行过程中,反应装置内的温度维持-60℃外,其余均与实施例1相同。
[0102]
本实施例中,由于反应装置内的温度过低,出现溶剂结块的问题,三甲硅烷基胺无法合成。
[0103]
实施例12本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,除步骤(1)进行过程中,反应装置内的温度初始温度为10℃外,其余均与实施例1相同。
[0104]
本实施例中,由于反应装置内的温度较高,副反应较多,仅少部分dsa转化为tsa。
[0105]
实施例13本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,除了将步骤(1)中的正壬烷等质量替换为正辛烷外,其余均与实施例1相同。
[0106]
实施例14本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,除了将步骤(1)中的正壬烷等质量替换为甲苯外,其余均与实施例1相同。
[0107]
实施例15
本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,除了将步骤(1)中的正壬烷等质量替换为质量比1:1的甲苯与正壬烷的组合外,其余均与实施例1相同。
[0108]
实施例16本实施例提供了一种利用副产物再生三甲硅烷基胺的方法,除了将步骤(1)中的正壬烷等质量替换为二甲苯外,其余均与实施例1相同记录实施例1-16提供方法的三甲硅烷基胺合成、纯化所用的时间,并测定最终所得三甲硅烷基胺的纯度。其中纯度采用气相色谱法进行分析,所得结果如表1所示。
[0109]
表1综上所述,本发明提供的方法以生产tsa过程中的副产物dsa为原料,配合对tsa生产过程中条件的调控,经氯硅烷化、氨置换及分离纯化,能够安全、稳定以及可靠的生产tsa,避免了dsa作为危废的浪费,从而使该反应装置生产tsa的过程符合绿色化工的要求,降低了环境风险,具有工业应用价值;本发明提供的利用副产物再生三甲硅烷基胺的反应装置,不仅能够实现dsa的充分利用,还能够稳定安全的实现tsa的生产,实现了对传统氯硅烷与氨制备tsa合成路线的改进,具有工业应用价值。
[0110]
以上所述仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,所属技术领域的技术人员应该明了,任何属于本技术领域的技术人员在本发明揭露的技术范围
内,可轻易想到的变化或替换,均落在本发明的保护范围和公开范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。