1.本发明属于纳米银制备技术领域,具体涉及一种3d打印的微流控反应装置及使用该装置合成形貌可控的纳米银的方法。
背景技术:
2.纳米粒子(nanoparticles,nps)是指在三维空间中至少一维处于纳米尺寸的微粒。由于小尺寸效应,nps具有独特的光、热、电、磁等物理和化学性质,在催化、导电和化学传感等领域得到了广泛应用。nps的化学组成、表面性质、形貌粒径、晶体构型、分散程度都决定了其应用性能。因此,精确控制合成过程中的每一步骤至关重要。然而,常规的批合成方法存在着粒径、形貌和质量的重现性差、无法实时监控合成过程、优化合成装置和条件的成本高等缺点,不利于纳米材料性能的提升。与传统的反应器相比,微流控体系的合成方法具有以下突出优势:(1)比表面积大的微通道可以加强传质传热速率,合成的产率更高,副反应少;(2)易精确控制合成条件、实现原位监测且试剂消耗量少;(3)多通道并行可成倍地提高反应通量,易扩展到实际生产。因此,与常规批合成方法相比,基于微流体装置的合成方法有望更精确地调控所合成nps的大小、组成、形态和粒径分布,并进行高通量智能化筛选合成。虽然基于层流微流体芯片的合成技术较为成熟,但其混合效率较低且抛物型的流体易造成通道接触面产生沉淀而堵塞通道。基于微流控技术生成的微液滴作为一个封闭的反应容器,液滴的四周都是不相溶的流体,减少了反应物的浓度弥散,使液滴内的固体反应产物不会与通道接触,因此,既不会发生通道的堵塞,又防止了交叉污染,并具有更窄的粒径分布范围。此外,液滴内部的循环流也提高了混合效率,缩短了反应时间。通过调控两相流速和合成温度,即可合成不同形貌、尺寸和粒径分布的nps,已被应用到磁球、硅球、中空微球、量子点、金属有机框架化合物、janus和贵金属等nps的合成。
3.据统计,2015年全球有1600余种商品中添加了nps,其中约1/4的产品明确表明含有抗菌性优良的ag nps。目前已报道t型和流动聚焦型结构的聚二甲基硅氧烷(polydimethylsiloxane,pdms)芯片设计,通过产生单分散液滴合成ag nps。然而,pdms芯片所依赖的传统模塑法,其工艺较为复杂且设备成本较高,越来越多的研究者开始直接利用商品化配件的微通道进行微流体装置的拼接,例如聚全氟乙丙烯管和三通接口、石英毛细管和聚四氟乙烯管,但是这些装置都依赖于购买到合适尺寸的商品化部件。3d打印是一种以计算机三维设计数字模型文件为基础,运用特殊可粘合材料逐层打印且快速成形构造物体的新技术。该技术最突出的优点是无需任何模具或过多机械修饰,只需在设计完成后就能直接生成任意形状的物体,相比于其它微加工技术,极大地降低了微流控芯片的技术门槛和加工成本,满足个性化定制的需求。在借助微流控技术合成单分散nps领域也有了一些初步的应用,例如,vini singh在《3d-printed device for synthesis of magnetic and metallic nanoparticles》(journal of flow chemistry,2021,11:135-142)中公开了一种低成本且可生物降解的3d打印设备,利用熔融沉积成型(fdm)工艺设计制作了基于t型通道的3d打印芯片,以十八烯作为油相,以还原剂柠檬酸钠和前驱体agno3作为水相,得
到了粒径为4.1
±
1nm的ag nps。但是其制备的ag nps的多分散指数(polydiseperse index,pdi)为0.06,粒径分布的均一度还有待改善。
技术实现要素:
4.针对上述问题,本发明提供了一种3d打印的微流控反应装置,及使用该装置合成形貌可控的纳米银(ag nps)的方法。该方法以agno3作为水相,液体石蜡作为油相,nabh4作为还原剂,pvp作为保护剂,通过优化各种反应参数以适配该微流控反应装置,实现了精确控制ag nps的形貌及粒径分布的目的。
5.为了实现上述目的,本发明具体采用如下技术方案:
6.一种微流控反应装置,所述微流控反应装置包含第一管道、第二管道、第三管道、第四管道、第五管道、第六管道、第七管道和第八管道;所述第一管道的一端与所述第二管道的一端均与所述第三管道垂直连通,且所述第一管道与所述第二管道同轴设置;所述第三管道一端与所述第四管道的一端连通,另一端与所述第五管道的一端连通;所述第四管道的另一端和所述第五管道的另一端均与所述第六管道的一端连通,所述第六管道的另一端为第一入口;所述第七管道的一端为第二入口,另一端与所述第二管道的另一端连通;所述第八管道的一端为第三入口,另一端与所述第二管道的另一端连通;所述第一管道的另一端为出口。
7.优选的,所述第一管道、第二管道、第三管道的内径均为0.8~1.4mm;所述第一管道、第二管道、第三管道的连通处构成“十”字型结构,且所述第一管道、第二管道、第三管道均在连通处向所述“十”字型结构的交叉点收缩,每个管道的内径均由0.8~1.4mm收缩至0.6~1.2mm,收缩的长度为0.3~10mm;进一步优选每个管道的内径均有0.8mm收缩至0.6mm,收缩的长度为0.3mm。
8.优选的,所述第一管道的长度为2~10mm;进一步优选所述第一管道的长度为6mm。
9.优选的,所述第六管道、第七管道、第八管道的内径均为0.8~1.4mm;进一步优选所述第六管道、第七管道、第八管道的内径均为0.8mm。
10.优选的,所述第四管道、第五管道的内径均为0.8~1.4mm;进一步优选所述第四管道、第五管道的内径均为0.8mm。
11.使用上述装置合成形貌可控的纳米银的方法,包括以下步骤:
12.s1、配制反应溶液:配制含5v/v%span 80的石蜡作为油相;以不含溶解氧的冰水配制nabh4溶液作为水相i;以含0.025mol/l edta的超纯水配制含agno3和pvp的溶液,然后调节溶液ph=10,作为水相ii;
13.s2、室温下,分别用三个注射器吸取油相、水相i、水相ii后置于三个注射泵上,将含油相的注射器、含水相i的注射器、含水相ii的注射器分别与所述微流控反应装置的第一入口、第二入口、第三入口相连接;启动注射泵,控制油相、水相i、水相ii的流量,待生成稳定的液滴后,将收集管置于所述微流控反应装置的出口处收集生成的液滴;
14.s3、关闭注射泵,将步骤s2中收集的液体在8000~11000rpm速度下离心5~20min,倒掉上层油相后加入丙酮;重复前述离心、加丙酮的步骤若干次,最后一次离心倒掉油相后加入超纯水,得到纳米银溶液。
15.优选的,所述水相ii中pvp的浓度为0.30~4.5g/l;进一步优选所述水相ii中pvp
的浓度为0.30~3.0g/l,最优选为3.0g/l。
16.优选的,nabh4与agno3的摩尔比值n的范围为:2≤n<12;进一步优选nabh4与agno3的摩尔比值n的范围为:2≤n≤8,最优选n=8。
17.优选的,所述油相的流量为150~1000μl/min,所述水相i的流量为3~10μl/min,所述水相ii的流量为3~10μl/min;进一步优选所述油相流量为500~1000μl/min,所述水相i的流量为5~10μl/min,所述水相ii的流量为5~10μl/min;最优选所述油相流量为1000μl/min,所述水相i的流量为10μl/min,所述水相ii的流量为10μl/min。
18.与现有技术相比,本发明的有益效果是:
19.(1)本发明中通过光固化(sla)3d打印机制作微流控反应装置,与fdm工艺相比,打印精度更高,速度更快,制作的微流控反应装置具有“十”字型结构,其“十”字交叉处的流动聚焦结构比t型结构具有更好的调控液滴尺寸和均一度的性能。
20.(2)本发明中还通过comsol多物理场耦合模拟软件对流动聚焦处的微通道结构进行流体动力学仿真,建立模拟微滴生成过程的模型,优化了微流控装置的结构尺寸。
21.(3)本发明中以前驱体agno3作为水相,液体石蜡作为油相,nabh4作为还原剂,pvp作为保护剂,使用上述微流控装置合成纳米银(ag nps);在合成过程中,通过调整各种参数(如pvp浓度、油相和水相流速等),得到与该微流控装置相适配的合成条件,实现了精确控制ag nps的形貌及粒径分布,制备的ag nps的pdi为0.01,粒径分布的均一性非常好。
附图说明
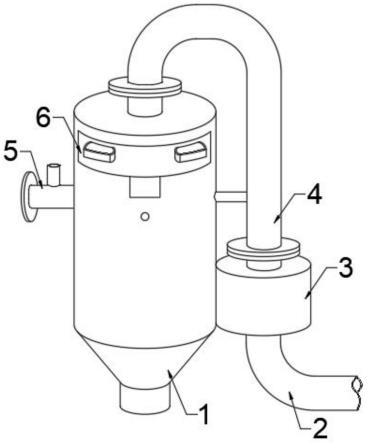
22.图1为实施例1的微流控反应装置;
23.图2为实施例2中基于comsol多物理场耦合模拟软件建立的液体微滴生成过程的模型图;
24.图3为实施例2中仿真模拟模型的液滴生成过程相场分布图;
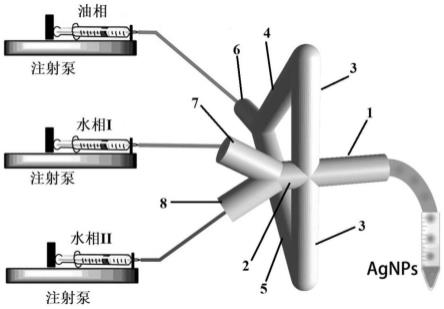
25.图4为实施例3中合成ag nps的过程示意图;
26.图5为实施例3中不同pvp浓度下合成的ag nps的uv-vis图;
27.图6为实施例3中不同nabh4/agno3摩尔比下合成的ag nps的uv-vis图;
28.图7为实施例3中不同油相、水相流量下合成的ag nps的uv-vis图;
29.图8为实施例3中不同油相、水相流量下合成的ag nps的sem图;
30.图9为实施例3中不同油相、水相流量下合成的ag nps的粒径分布直方图;
31.图10为实施例3中不同油相、水相流量下合成ag nps时液滴生成的仿真图;
32.图11为实施例3中第12号样品的hrtem图;
33.图12为实施例3中第12号样品元素分析结果图;
34.图13为对比例中合成ag nps的几何模型图;
35.图14为对比例中合成ag nps时的仿真模拟模型的液滴生成过程的相场分布图。
36.图中:1、第一管道;2、第二管道;3、第三管道;4、第四管道;5、第五管道;6、第六管道;7、第七管道;8、第八管道。
具体实施方式
37.以下结合实施例对本发明技术方案进行清楚、完整的描述,显然,所描述的实施例
仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施例,都属于本发明所保护的范围。本领域技术人员依据以下实施方式所作的任何等效变换或替代,均属于本发明的保护范围之内。
38.实施例1
39.本实施例通过3d打印制作了一种微流控反应装置,具体地,先通过三维cad软件autodesk123d设计该微流控装置的结构,并转换为stl文件格式;再利用sla 3d打印机打印出所设计的微流控反应装置,打印参数设置为激光波长355nm,激光能量1000mw,激光扫描速度2m/s,层厚0.1mm/layer。打印出的微流控反应装置如图1所示,包括第一管道1、第二管道2、第三管道3、第四管道4、第五管道5、第六管道6、第七管道7、第八管道8。第一管道1的一端与第二管道2的一端均与第三管道3垂直连通形成“十”字型结构,即第一管道1与第二管道2同轴设置,第一管道1、第二管道2、第三管道3的轴线相交于同一平面内。第一管道1的另一端为出口。第三管道3的一端与第四管道4的一端连通,另一端与第五管道5的一端连通。第四管道4的另一端和第五管道5的另一端均与第六管道6的一端连通,第六管道6的另一端为第一入口。第七管道7的一端为第二入口,另一端与第二管道2的另一端连通。第八管道8的一端为第三入口,另一端与第二管道2的另一端连通。
40.实施例2
41.为了研究微流控反应装置对合成ag nps时液滴的性能调控效果最好的尺寸(如微流控通道的长度、深度、宽度等),本实施例基于comsol多物理场耦合模拟软件对流动聚焦处的微通道(即微流控反应装置的各种管道)结构进行流体动力学仿真,建立模拟液体微滴生成过程的模型,通过模拟的数据指导3d打印的微流控反应装置选择最适合的尺寸。
42.仿真模拟时实际流体的性质参数(包括黏度、密度等)列于表1。考虑壁面润湿性对分散相的影响,先以相同sla技术经过相同步骤制作出方块,如图2所示,测定其接触角为85
°
,所以将计算方程中代表微通道壁面润湿性的接触角参数设定为85
°
。由于通道内径越小越易调控液滴尺寸,考虑到sla 3d打印技术的发展现状,为了保证通道通畅且满足精度和机械强度,所有管道的内径都至少为0.8mm,壁厚都至少为0.8mm。当管道内径超过1.4mm时,微流控反应装置的骤窄交叉处不同方向的内部通道会重叠,无法分别引入油水两相,因此设定管道的内径为0.8~1.4mm。此外,流动聚焦处的骤窄结构设计有利于增加连续相的剪切力,因此,根据上述尺寸建立如图2所示的几何模型,油相作为连续相流体从第三管道3(内径为0.8mm)的两端流入;两个水相混合后作为分散相由第二管道2(内径为0.8mm)流入;第一管道1、第二管道2、第三管道3的连通处为“十”字型结构的流动聚焦处,该流动聚焦处设置成骤窄的结构,该骤窄结构的交叉处(即“十”字型结构的交叉处)的截面由四个相同的虚拟的等腰梯形(由于管道是中空结构,因此实际上每个管道在交叉处的截面的形状不是完整的等腰梯形,其缺少实际的顶边和底边,故称之为“虚拟的等腰梯形”)构成,每个等腰梯形的顶边长0.6mm,底边长0.8mm,高为0.3mm;即截面为第一管道1、第二管道2、第三管道3各自的轴线相交的平面,等腰梯形的底边长度即为与之对应的管道的内径长度,第一管道1和第二管道2均向“十”字型结构的交叉处收缩,二者的收缩长度均为等腰梯形的高度,二者收缩至交叉处的管道内径均为等腰梯形的顶边长度;第三管道3从两端向“十”字型结构的交叉处收缩,每端的收缩长度为等腰梯形的高度,每端收缩至交叉处的管道内径均为等腰
梯形的顶边长度。生成的液滴从第一管道1流出被收集于ep管内。
43.表1用于仿真模型的参数设定
[0044][0045][0046]
按照上述设置得到如图3所示的相场分布,完整记录了第一个液滴的生成过程,十字型通道内液滴的生成过程可以为液滴生长和从颈部分裂两个阶段。第一阶段为(a)到(g),持续从上下挤压的连续相使从左端进入交叉处的分散相前端逐渐发生形变,颈部不断变细。图3中(h)到(i)为液滴生成第二阶段,由于连续相黏性剪切力的持续作用,表面张力不能够维持相界面的形状,到5.608s时液滴在连续相剪切力的作用下脱离,形成单个液滴。由图3中(h)和(i)可知,第一管道1的长度为2mm以上,足以保证生成稳定的液滴。考虑到套入泵管的密封性和操作性,将第一管道1的长度设定为6mm。当第一管道1的长度超过10mm时,容易导致内部通道中未光固化的液态光敏树脂难以清洗,因此将第一管道的长度设定为2~10mm。
[0047]
实施例3
[0048]
本实施例中使用实施例1提供的装置合成ag nps,根据实施例2的研究将所有管道的内径设置为0.8mm,第一管道1的长度设置为6mm,“十”字交叉结构的骤窄处的截面由四个相同的等腰梯形构成,每个等腰梯形的顶边长0.6mm,底边长0.8mm,高为0.3mm。具体的制备方法包括以下步骤:
[0049]
s1、配制反应溶液:配制含5v/v%span 80的石蜡作为油相;以不含溶解氧的冰水配制nabh4溶液作为水相i;以含0.025mol/l edta的超纯水配制含agno3和pvp(聚乙烯吡咯烷酮)的混合溶液,然后以0.1mol/l氢氧化钠溶液调节混合溶液ph=10,作为水相ii;
[0050]
s2、分别用三个注射器吸取油相、水相i、水相ii后置于三个注射泵上,将含油相的注射器、含水相i的注射器、含水相ii的注射器分别与微流控反应装置的第一入口、第二入口、第三入口相连接;启动注射泵,控制油相、水相i、水相ii的流量,待生成稳定的液滴后,将ep管置于微流控反应装置的出口处收集生成的液滴(如图4所示);
[0051]
s3、当ep管中液体体积达到1ml时,关闭注射泵,将步骤s2中收集的液体在8000~11000rpm速度下离心5~20min,倒掉上层油相后加入丙酮;重复前述离心、加丙酮的步骤3次,最后一次离心倒掉油相后加入10ml超纯水,得到ag nps溶液。
[0052]
装置使用结束后,管道内可能会有反应物残留,影响下一次产物合成,因此需要进行清洗,具体清洗步骤如下:借助泵管将装有超纯水或乙醇的注射器与微流控装置的各个入口相连接,开启注射泵,将水或乙醇泵入微流控装置,清洗内部通道3遍以上,最后使用氮气吹干内部通道。
[0053]
本实施例通过单因素实验研究了影响ag nps合成的非结构类因素,主要研究因素包括pvp含量、nabh4/agno3摩尔比、油相和水相的流量。对合成的ag nps进行uv-vis(紫外-可见)吸收光谱和sem(扫描电子显微镜)测定。通过uv-vis吸收峰位置可以看出最大吸收波
长(λm)是否在纳米粒子(nps)的特征范围内,进而判定其是否符合典型nps溶胶所特有的光学特征,即λm在可见光区产生表面等离子共振吸收。通过吸收峰强度可以看出其最大吸光度(am),因为nps浓度(即产率)和am正比例关系,进而反映出nps溶液的浓度,即单位体积内的粒子数。根据吸收峰形态可以估算出吸收光谱的半峰宽fwhm(full width at half maximum,半峰宽指最大吸光度一半的两点处对应的波长差),通过半峰宽可以反映nps的粒度分布情况,半峰宽数值越小说明粒子粒径均匀性更好。还可以根据吸收峰的位置变化来反映nps平均粒径的变化,最大吸收波长λm向短波移动为蓝移,说明平均粒径变小;相反,红移说明平均粒径变大。
[0054]
1、pvp含量
[0055]
在nabh4/agno3摩尔比为4:1、油相流量q
oil
=150μl/min、水相i流量q
w1
=3μl/min、水相ii流量q
w2
=3μl/min的条件下,研究不同pvp浓度下合成的ag nps的形貌,pvp浓度和实验结果见表2,不同pvp浓度下合成的ag nps的uv-vis图见图5。
[0056]
表2不同pvp浓度时合成ag nps的实验结果
[0057][0058]
从表2中可以看出,pvp浓度与ag nps的形核、生长过程有密切关系,随着pvp浓度的增加,紫外可见(uv-vis)吸收光谱的半峰宽fwhm越来越小,说明所合成的ag nps的粒度分布越来越均匀。除了1号样品外,其它样品的λm和am大致相同,说明pvp浓度达到一定程度后对ag nps粒径和数量的影响不大;但是当pvp浓度大于4.5g/l时,部分ag nps形貌呈现出正方体。这主要是因为pvp是不带电荷的非离子表面活性剂,通过n和o原子与银晶核配位形成络合物保护层,留下疏水性碳链伸向四周阻止了颗粒间的相互聚合团聚,从而达到防止晶粒长大和降低粒子团聚的作用;但是pvp浓度太大时,pvp将有足够大的趋势与(111)晶面结合,使(111)晶面也受到钝化而无法正常生长。根据图5可知,uv-vis吸收光谱的λm均在400~420nm,符合球形ag nps所特有的光学特征,因此加入pvp后易于合成球形或类球形ag nps。这是由于羰基的存在,可以氢键化溶剂分子,从而与晶体相结合并包裹在晶体周围,让晶体呈现一定形貌来生长,起到生长改性剂的作用。此外,吸附了pvp分子的银晶核的表面结合能降低,使反应后继续生成银原子在晶核表面均匀生长。为了生成单分散性最好的球形ag nps,后续研究中选择pvp浓度为3g/l。
[0059]
2、nabh4/agno3摩尔比
[0060]
在pvp浓度c
pvp
=3g/l、油相流量q
oil
=150μl/min、水相i流量q
w1
=3μl/min和水相
ii流量q
w2
=3μl/min条件下,研究不同nabh4/agno3摩尔比合成的ag nps的形貌,nabh4/agno3摩尔比和实验结果见表3,不同nabh4/agno3摩尔比下合成的ag nps的uv-vis图如图6所示。
[0061]
表3不同nabh4/agno3摩尔比时合成ag nps的实验结果
[0062][0063]
由表3可知,当还原剂nabh4浓度较低时,还原速率过慢,混合初期反应体系中合成的银原子浓度低,难以达到形成银晶核的过饱和度;由于形成的晶核数量少,同时体系中被还原析出的银原子数量少,形成的ag nps的直径较小且粒径均一性较差。当nabh4/agno3摩尔比值n≥2时,随着还原剂浓度的增加,在成核阶段溶液中生成了数量较多的银原子,很快达到晶核的过饱和度,形成了较多的银晶核,使合成ag nps的粒径越大,单位体积的数量也越多即ag nps数量溶液浓度越大。当nabh4/agno3摩尔比值n=12时,还原速率过快,导致银晶核间碰撞机率加大,易发生团聚,使ag nps的粒径及其分布都增大。从图6中可以看出,所合成ag nps的λm最大吸收波长均在410~420nm区间,符合球形ag nps的光学特征。为了使反应体系中形成的银晶核数量与晶核生长过程中所需的银原子数量相对平衡,合成单分散性较好的球形或类球形的30nm ag nps,后续研究中选择nabh4/agno3摩尔比值n=8。
[0064]
3、油相和水相的流量
[0065]
在pvp浓度c
pvp
=3g/l、nabh4/agno3摩尔比值n=8的条件下,研究不同的油相、水相流量合成的ag nps,油相和水相流量及实验结果见表4,合成的ag nps的uv-vis图、sem图、粒径分布直方图以及液滴生成仿真图如图7~10所示,图8~10中各个图上的9、10、11、12分别对应表4中的样品编号。
[0066]
表4不同油相、水相流量合成ag nps的实验结果
nanoparticles》中公开的t型微流控反应装置的尺寸建立如图13所示的几何模型,每个通道的内径均为3mm;采用本发明中的油相作为连续相从t型结构的垂直端流入,水相i和水相ii分别从另外两个水平端入口流入后形成的混合液作为分散相;油相、水相i、水相ii的组成以及三者的流量均与实施例3中第12号样品保持一致;得到的相场分布如图14所示。从图14中可以看出,该装置中的油相在较长时间内(长达500s)都无法将水相剪切生成单分散液滴,因此无法采用本发明的方法在该装置中成功合成ag nps。可见,本发明中合成ag nps的方法与本发明中的微流控装置配合使用才能达到更好的调控ag nps形貌的效果,该方法与现有技术中的微流控装置的适配性明显不如本发明中的装置。
[0076]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明的保护范围。对于任何熟悉本领域的技术人员来说,本发明可以有各种更改和变化。任何依据本发明申请保护范围及说明书内容所作的简单的等效变化和修饰,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。