1.本发明涉及抑制剂加工技术领域,具体为一种抑制剂及其制备方法。
背景技术:
2.hq461(cas:1226443-41-9)是一种促进cdk12-ddb1相互作用以触发细胞周期蛋白k降解的分子胶,在专利wo2021249517a1(page26)中公开了其合成方法:首先通过5-甲基-2-氨基噻唑(a)氨解原料b得到化合物c,c通过溴素溴化得到化合d,d与化合物e关环得到hq461。
3.其路线中第二步用到溴参与反应,且需要在60℃温度下滴加参与反应,对反应设备及操作要求较高,此外该反应过程中副产物多,纯化用到柱层析,提纯繁琐,不易于放大量产。
技术实现要素:
4.发明的目的针对上述技术问题,本发明提供了一种抑制剂及其制备方法,用以解决背景技术中提到的现有的hq461合成反应过程中副产物多,纯化用到柱层析,提纯繁琐,不易于放大量产的技术问题。
5.技术方案为了达到上述目的,本发明提供的技术方案为:一种抑制剂的制备方法,包括以下步骤:步骤1:将n-(6-甲基-2-吡啶基)硫脲e及4-氯乙酰乙酸乙酯加入溶剂中回流反应2小时,反应生成中间体f;步骤2:将中间体f水解生成中间体g;
步骤3:将中间体g与5-甲基-2-氨基噻唑(a)缩合得到hq461。
6.进一步的改进在于:步骤1中,n-(6-甲基-2-吡啶基)硫脲及4-氯乙酰乙酸乙酯摩尔比为1:(1-3),优选1:1.2。
7.进一步的改进在于:步骤1中,所述溶剂选自无水甲醇,无水乙醇及四氢呋喃中的至少一种,优选无水乙醇。
8.进一步的改进在于:步骤1的反应温度25-80℃,优选70-80℃。
9.进一步的改进在于:步骤2中,所述水解试剂选自氢氧化钠,氢氧化钾和氢氧化锂中的至少一种。
10.进一步的改进在于:步骤2中,所述水解试剂为氢氧化钠,氢氧化钠摩尔比为1:(1-10),优选1:3。
11.进一步的改进在于:步骤2中,所述溶剂选自无水甲醇,无水乙醇及四氢呋喃中的至少一种,优选无水甲醇。
12.进一步的改进在于:步骤2的反应温度25-80℃,优选60℃。
13.进一步的改进在于:步骤3中,所述缩合剂选自hatu,edci,t3p中的至少一种。
14.进一步的改进在于:步骤3中,所述缩合剂选自为t3p,t3p摩尔比为1:(1-3),优选1:1.2。
15.进一步的改进在于:步骤3中,所述溶剂选自二氯甲烷,乙腈,四氢呋喃及dmf的至少一种,优选dmf。
16.进一步的改进在于:步骤3的反应温度0-80℃,优选10-30℃。
17.本发明还公开了一种抑制剂,其采用上述制备方法制得。
18.有益效果本发明提供的技术方案,与现有技术相比,具有以下有益效果:本发明的合成方法,第一步中间体粗品直接用于下步反应,通过前两步反应合并纯化,减少纯化步骤,提高合成效率,同时避免了原合成工艺里易制毒溴素的使用。
19.工艺操作简单高效,所有中间体及产品均未用柱层析分离,可通过打浆直接得到纯品,提高了操作简易性。
具体实施方式
20.下面结合具体实施例对本发明的技术方案作进一步的描述,但本发明并不限于这些实施例。
实施例
21.需要说明的是,1h nmr图谱是用bruker仪器 (400mhz)测定而得,化学位移用ppm表示。使用四甲基硅烷内标准(0 .00ppm)。1h nmr的表示方法:s=单峰,d=双重峰,t=三重峰,m=多重峰,br=变宽的,dd=双重峰的双重峰,dt= 三重峰的双重峰。若提供偶合常数时,其单位为hz。
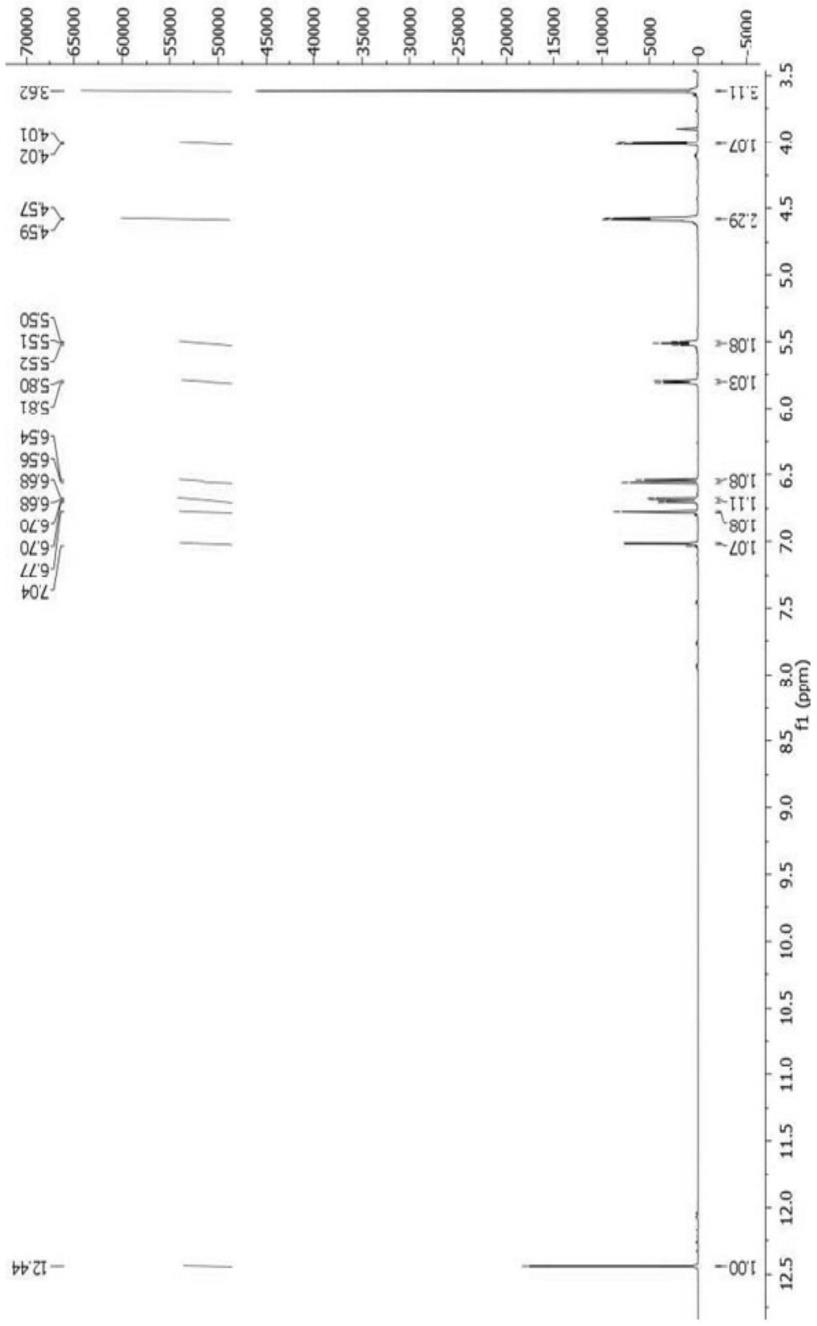
22.质谱是用lc/ms仪测定得到,离子化方式可为esi或apci。
23.该hq461的合成方法如下:第一步:将n-(6-甲基-2-吡啶基)硫脲(10g,59.80mmol,1eq)及4-氯乙酰乙酸乙酯(11.8g,71.76mmol,1.2eq)加入无水乙醇(100ml)中回流反应2小时。反应液冷却,减压浓缩除去乙醇,浓缩剩余物直接用于下步反应。
24.第二步:上步所得产品粗品加入甲醇(100ml),h2o(100ml)及naoh(7.2g,179.39mmol,3eq)于60℃反应1小时后,反应液冷却浓缩除去甲醇,剩余物于冰浴下用2n hcl调ph至4-5,有固体析出,抽滤,固体用水及mtbe打浆后烘干得产品10 g(灰白色固体)。
25.其中,两步合并产率67.11%。
26.1 h nmr(400mhz ,dmso-d6)δ12.30(br ,1h),11.23(br ,1h),7.54-7.58(t ,1h) ,6.79-6.81(d ,1h), 6.75-6.76(d ,1h),6.72(s ,1h), 3.55(s ,2h), 2.44(s ,3h)第三步:反应瓶中加入上步所得产品(10g,40.11mmol,1eq),5-甲基-2-氨基噻唑(4.6g,40.11mmol,1eq),t3p(30.6g,48.13mmol,1.2eq),dipea(10.34 g,80.22mmol,2eq) 于dmf中,室温下反应2小时。反应液加水,ea萃取,合并ea相,用饱和nacl洗,ea相用无水硫酸钠干
燥,过滤,滤液浓缩,剩余物用乙腈打浆得6.3g产品hq461(产率y:45.48%)。
27.1 h nmr(400mhz ,dmso-d6)δ12.05(br ,1h),11.23(br ,1h),7.53-7.57(t ,1h), 7.13(s ,1h) ,6.79 (s ,1h), 6.76-6.77(d ,1h),6.75(s ,1h), 3.75(s ,2h), 2.44(s ,3h), 2.33(s ,3h)lcms:m/z=346(m 1)尽管已用具体实施方案来说明和描述了本发明,但对于本领域的技术人员显而易见的是,在不背离本发明的精神和保护范围的情况下可作出许多其它的变化和修改。因此,有意识地在附加的权利要求书中包括属于本发明范围内的所有这些变化和修改。
技术特征:
1.一种抑制剂的制备方法,其特征在于,包括以下步骤:步骤1:将n-(6-甲基-2-吡啶基)硫脲及4-氯乙酰乙酸乙酯加入溶剂中回流反应2小时,反应生成中间体f;步骤2:将中间体f水解生成中间体g;步骤2:将中间体f水解生成中间体g;步骤3:将中间体g与5-甲基-2-氨基噻唑缩合得到hq461。2.根据权利要求1所述的一种抑制剂的制备方法,其特征在于:步骤1中,n-(6-甲基-2-吡啶基)硫脲及4-氯乙酰乙酸乙酯摩尔比为1:(1-3)。3.根据权利要求1所述的一种抑制剂的制备方法,其特征在于:步骤1中,所述溶剂选自无水甲醇,无水乙醇及四氢呋喃中的至少一种。4.根据权利要求1所述的一种抑制剂的制备方法,其特征在于:步骤1的反应温度25-80℃。5.根据权利要求1所述的一种抑制剂的制备方法,其特征在于:步骤2中,所述水解试剂选自氢氧化钠,氢氧化钾和氢氧化锂中的至少一种。6.根据权利要求1所述的一种抑制剂的制备方法,其特征在于:步骤2中,所述溶剂选自无水甲醇,无水乙醇及四氢呋喃中的至少一种。7.根据权利要求1所述的一种抑制剂的制备方法,其特征在于:步骤2的反应温度25-80℃。8.根据权利要求1所述的一种抑制剂的制备方法,其特征在于:步骤3中,所述缩合剂选自hatu,edci,t3p中的至少一种。9.根据权利要求1所述的一种抑制剂的制备方法,其特征在于:步骤3中,所述溶剂选自二氯甲烷,乙腈,四氢呋喃及dmf的至少一种。
10.一种抑制剂,其特征在于:该抑制剂采用权利要求1-9任一所述的制备方法制得。
技术总结
本发明涉及一种抑制剂的制备方法,包括以下步骤:步骤1:将N-(6-甲基-2-吡啶基)硫脲及4-氯乙酰乙酸乙酯加入溶剂中进行环化反应,减压浓缩除去溶剂,得中间体F粗品直接用于下步反应;步骤2:将中间体F水解,用水及MTBE打浆后烘干得产品中间体G;步骤3:将中间体G与5-甲基-2-氨基噻唑通过缩合反应得到HQ461。本发明的合成方法第一步中间体粗品直接用于下步反应,通过前两步反应合并纯化,减少纯化步骤,提高合成效率,同时避免了原合成工艺里易制毒溴素的使用,且本发明工艺操作简单高效,所有中间体及产品均未用柱层析分离,可通过打浆直接得到纯品,提高了操作简易性。提高了操作简易性。
技术研发人员:袁钧 孟朵 朱成智
受保护的技术使用者:安庆研至医药科技有限公司
技术研发日:2022.08.10
技术公布日:2022/12/12
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。