1.本发明属于有机合成技术领域,具体涉及一种铜氧化n,s-缩烯酮类化合物环化成异噻唑啉酮类化合物的合成方法。
背景技术:
2.异噻唑啉酮类化合物是一种广谱、高效、低毒、非氧化性杀生剂,可以杀灭细菌、真菌、霉菌及霉菌,其杀菌原理主要依靠杂环上的活性部分破坏菌体内的dna分子,使菌体失去活性,具有杀生效率高,降解性好,具有不产生残留、操作安全、配伍性好、稳定性强等特点。异噻唑啉酮类杀菌剂已经广泛运用于油田、造纸、农药、切削油、皮革、油墨、染料、制革等行业(z.k.yu,et al.chem.eur.j.2018,24,14368-14372.)。因此,研究开发一种反应条件温和、效率高的异噻唑啉酮类化合物的合成方法成为当前亟待研究的重要课题。
技术实现要素:
3.鉴于此,本发明的目的在于提供一种铜氧化n,s-缩烯酮类化合物环化成异噻唑啉酮类化合物的合成方法。为了实现上述目的,本发明的技术方案如下:
4.一种异噻唑啉酮类化合物,其分子结构式如下式所示:
[0005][0006]
r1、r2选自c
1-c
18
的烷基、含有取代基的c
1-c
18
的烷基、c
6-c
18
的芳基或含有取代基的c
6-c
18
的芳基,所述取代基为氟、氯、溴、碘、甲基、叔丁基、苯基、甲氧基、氰基、乙酰氧基中的一种或两种以上。
[0007]
上述技术方案中,进一步地,r1、r2选自c
1-c
18
的直链烷基、含有取代基的c
1-c
18
的直链烷基、c
6-c
18
的芳基或含有取代基的c
6-c
18
芳基,所述取代基为氟、氯、溴、碘、甲基、叔丁基、苯基、甲氧基、氰基、乙酰氧基中的一种,所述含有取代基的c
6-c
18
芳基为所述取代基在邻、间或对单取代的芳基。
[0008]
本发明的另一目的在于提供一种异噻唑啉酮类化合物的合成方法,以铜盐为氧化剂,在碱的条件下,在反应溶剂中n,s-缩烯酮类化合物(2)发生自身环化反应,反应结束后经常规分离纯化方法进行产物分离和表征,一步生成异噻唑啉酮类化合物(1);
[0009]
反应式如下所示:
[0010][0011]
上述技术方案中,进一步地,所述铜盐为溴化铜、氯化铜的一种或两种。
[0012]
上述技术方案中,进一步地,所述碱选自碳酸锂、碳酸钠、碳酸钾、醋酸钠、醋酸钾、醋酸锂、叔丁醇钠、叔丁醇钾或叔丁醇锂中的一种或两种以上的混合物。
[0013]
上述技术方案中,进一步地,所述n,s-缩烯酮类化合物(2)与氧化剂的摩尔比为10:1-1:10;n,s-缩烯酮类化合物(2)与碱的摩尔比为10:1-1:10。
[0014]
上述技术方案中,进一步地,所述反应溶剂为n,n-二甲基甲酰胺、二氯乙烷、二氧六环、吡啶、乙酸、苯的一种或两种以上的混合溶剂。
[0015]
上述技术方案中,进一步地,反应气氛为空气、氧气、氮气、氩气中的一种或两种。
[0016]
上述技术方案中,进一步地,反应温度为85-150℃,反应时间为0.5h-12h。
[0017]
上述技术方案中,进一步地,所述n,s-缩烯酮类化合物(2)与氧化剂的摩尔比为1:3。
[0018]
上述技术方案中,进一步地,所述n,s-缩烯酮类化合物(2)与碱的摩尔比为1:3。
[0019]
上述技术方案中,进一步地,所述反应温度为120℃。
[0020]
上述技术方案中,进一步地,所述反应时间为2h。
[0021]
和现有技术相比,本发明取得如下有益效果:本发明方法以n,s-缩烯酮类化合物为原料,利用n,s-缩烯酮类化合物在高温下发生铜氧化的自身环化反应合成不同类型的异噻唑啉酮类化合物,所得产物具有官能团多样性,制备的原料易得、操作简便、合成反应条件温和、效率高。为异噻唑啉酮类化合物在油田、造纸、农药、切削油、皮革、油墨、染料、制革等行业的广泛运用提供重要保障。
具体实施方式
[0022]
以下结合具体实施例对本发明做进一步说明。通过下述实施例有助于进一步理解本发明,但本发明的内容并不仅限于此。
[0023]
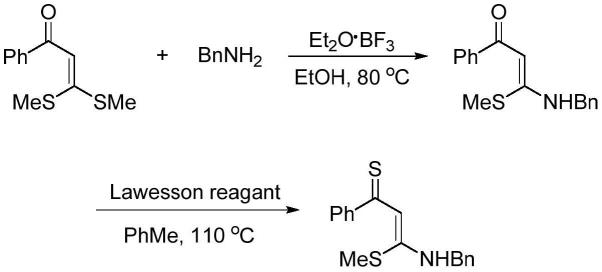
本发明从n,s-缩烯酮类化合物出发,合成异噻唑啉酮类化合物。下述实施例中原料(2a)制备的反应式如下,具体实验步骤参考文献(j.org.chem.2014,79,10553-10560)的方法来制备。
[0024][0025]
实施例1
[0026]
一种异噻唑啉酮类化合物的合成方法,反应式如下:
[0027]
[0028]
具体过程为:称取1-甲硫基-1-苄胺-1-丁烯-3-苯基-3-硫酮(2a)(90mg,0.3mmol)、溴化铜(201mg,0.9mmol)和叔丁醇锂(72mg,0.9mmol)于10ml反应管中,加入n,n-二甲基甲酰胺3ml,在120℃下搅拌反应2小时。反应结束后,减压下除去挥发组份,然后通过硅胶柱层析分离(洗脱液为石油醚(60-90℃)/乙酸乙酯,v/v=20:1),得到黄色液体目标产物(1a)(56mg,收率70%)。目标产物通过核磁共振谱测定得到确认。
[0029]
化合物表征数据
[0030]
异噻唑啉酮化合物(1a):黄色液体,1h nmr(400mhz,cdcl3)δ7.43(m,5h),7.36(m,5h),6.52(s,1h),4.98(s,2h).
13
c{1h}nmr(100mhz,cdcl3)δ169.24,156.37,136.38,131.01,130.30,129.37,129.00,128.53,128.43,125.89,110.12,47.49。
[0031]
实施例2
[0032]
一种异噻唑啉酮类化合物的合成方法,反应式如下:
[0033][0034]
反应步骤与操作同实施例1,与实施例1不同之处在于,氧化剂为氯化铜。停止反应,经后处理得到目标产物(1a)(51mg,收率63%)。
[0035]
实施例3
[0036]
一种异噻唑啉酮类化合物的合成方法,反应式如下:
[0037][0038]
反应步骤与操作同实施例1,与实施例1不同之处在于,碱为叔丁醇钠。停止反应,经后处理得到目标产物(1a)(39mg,收率49%)。
[0039]
实施例4
[0040]
一种异噻唑啉酮类化合物的合成方法,反应式如下:
[0041][0042]
反应步骤与操作同实施例1,与实施例1不同之处在于,碱为碳酸钾。停止反应,经后处理得到目标产物(1a)(46mg,收率57%)。
[0043]
实施例5
[0044]
一种异噻唑啉酮类化合物的合成方法,反应式如下:
[0045][0046]
反应步骤与操作同实施例1,与实施例1不同之处在于,碱为碳酸钠。停止反应,经后处理得到目标产物1a(37mg,收率46%)。
[0047]
实施例6
[0048]
反应步骤与操作同实施例1,与实施例1不同之处在于,1-甲硫基-1-苄胺-1-丁烯-3-苯基-3-硫酮(2a)与溴化铜的摩尔比为2.5:1。停止反应,经后处理得到目标产物1a(52mg,收率65%)。
[0049]
实施例7
[0050]
反应步骤与操作同实施例1,与实施例1不同之处在于,1-甲硫基-1-苄胺-1-丁烯-3-苯基-3-硫酮(2a)与叔丁醇锂的摩尔比为1:2。停止反应,经后处理得到目标产物(1a)(41mg,收率51%)。
[0051]
实施例8
[0052]
反应步骤与操作同实施例1,与实施例1不同之处在于,反应时间为1h。停止反应,经后处理得到目标产物(1a)(47mg,收率59%)。
[0053]
实施例9
[0054]
反应步骤与操作同实施例1,与实施例1不同之处在于,反应时间为1.5h。停止反应,经后处理得到目标产物(1a)(50mg,收率62%)。
[0055]
对于任何熟悉本领域的技术人员而言,在不脱离本发明技术方案范围情况下,都可利用上述揭示的技术内容对本发明技术方案作出许多可能的变动和修饰,或修改为等同变化的等效实施例。因此,凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所做的任何简单修改、等同变化及修饰,均应仍属于本发明技术方案保护的范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。