1.本发明属于有机合成领域,具体涉及一种吖啶四氟硼酸盐类光反应催化剂及其合成方法。
背景技术:
2.吖啶盐类化合物是一类重要的光催化剂结构,含有该类骨架的有机分子在光催化领域具有非常广泛的应用,因此探索高效经济的吖啶盐合成法一直备受关注。其中吖啶四氟硼酸盐类光反应催化剂作为一类常见的光催化剂广泛应用于多种碳氢键、碳氟键的活化,其参与的高效光催化反应改善了多种化合物的合成方法,为受电子效应局限化合物的合成提供了更多可能。[a)romero,n,a.,margrey,k,a.,tay,n,e.,nicewicz,d,a.,science.2015,349,1326.b)pistritto,v,a.,schutzbach-horton,m,e.,nicewicz,d,a.,j.am.chem.soc.2020,142,40.c)qi y.,jia z.,li l.,zhang l.,org.chem.front.2018,5,237.]。目前合成吖啶四氟硼酸盐的方法较少[d)hutskalova,v.,sparr c.,org.lett.2021,23,5143.e)sparr,c.,xing wu.,angew.chem.int.ed.2022,61,e202201424.],其中反应需要叠氮化物,操作危险,且反应环境-35℃,条件苛刻,反应步骤过多。因此,现有的制备吖啶四氟硼酸盐类光反应催化剂的工艺复杂,调条件苛刻,操作环境存在安全隐患。
技术实现要素:
[0003]
针对现有技术中存在的问题,本发明的目的在于提供一种吖啶四氟硼酸盐类光反应催化剂及其合成方法,本发明以亚胺类化合物为反应原料,在溶剂的存在下,单分子反应得到中间二氢吖啶,再经过渡金属催化偶联、氧化、阴离子交换高效合成吖啶四氟硼酸盐类光反应催化剂,有效的减少了反应步骤且操作环境存在安全制备得到的产物稳定,反应周期短,能高效得到目标产物;
[0004]
本发明是通过以下技术方案来实现:
[0005]
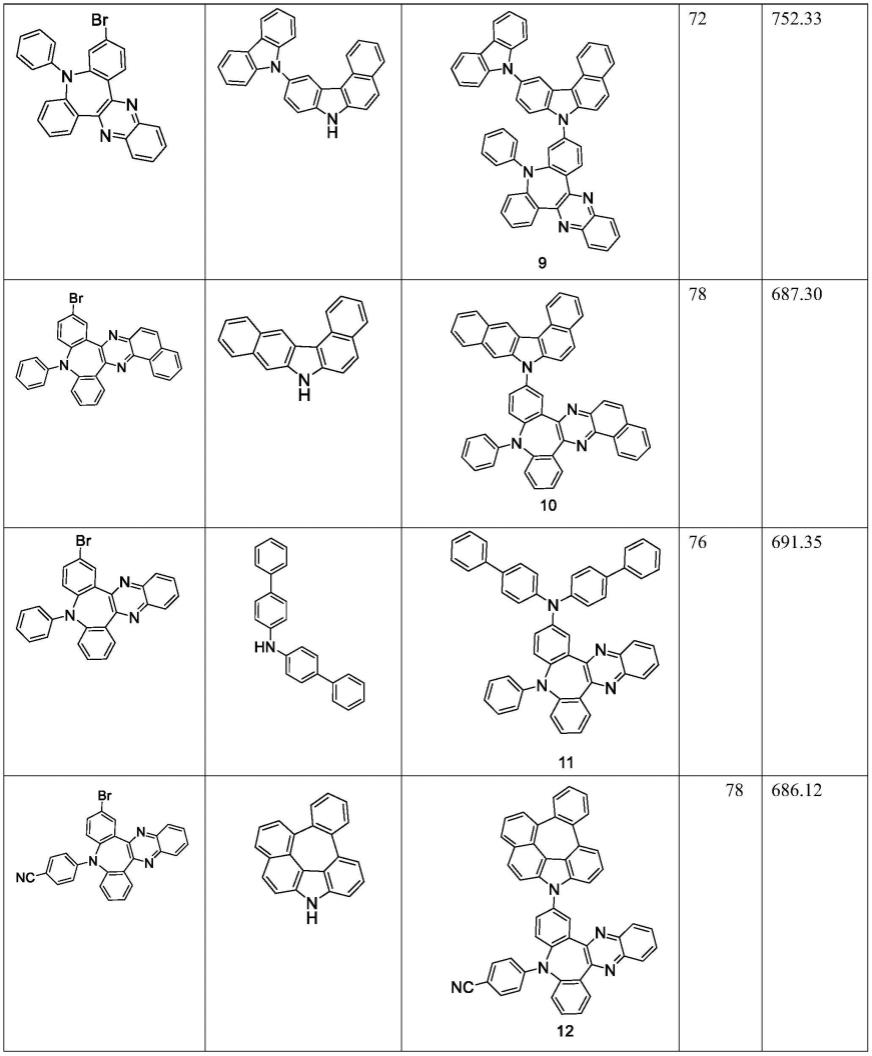
一种吖啶四氟硼酸盐类光催化剂,所述吖啶四氟硼酸盐类光催化剂的化学式为:
[0006][0007]
其中,r1选自氢、烷基、甲氧基、甲硫基、卤素、硝基或苯基;r2选自氢、烷基、杂环、或苯基;r3选自氢、烷基、甲氧基、甲硫基、酯基、卤素、杂环或硝基。
[0008]
一种吖啶四氟硼酸盐类光催化剂的合成方法,包括以下步骤;
[0009]
步骤1,在溶剂中加入将亚胺类化合物,经alcl3催化在惰性气体保护下进行反应后得到二氢吖啶类化合物;
[0010]
步骤2,将所述的二氢吖啶类化合物与卤代芳烃化合物通过过渡金属钯催化反应后得到n-芳基吖啶类化合物;
[0011]
步骤3,对所述的的n-芳基吖啶类化合物进行氧化后,再通过阴离子交换反应后制备得到吖啶四氟硼酸盐类光催化剂。
[0012]
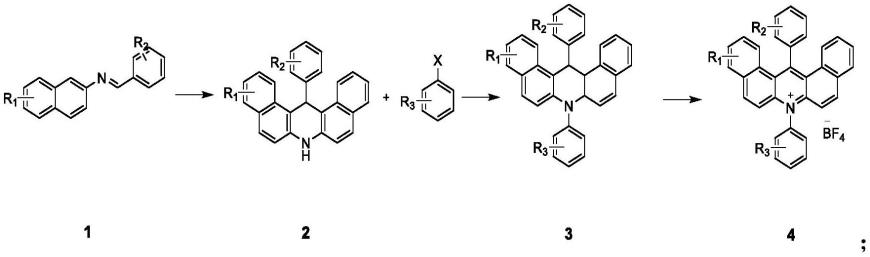
优选的,所述吖啶四氟硼酸盐类光催化剂的合成反应式为:
[0013][0014]
其中,r1选自氢、烷基、甲氧基、甲硫基、卤素、硝基或苯基;r2选自氢、烷基、杂环、或苯基;r3选自氢、烷基、甲氧基、甲硫基、酯基、卤素、杂环或硝基,x为卤素。
[0015]
优选的,所述步骤1中亚胺类化合物与alcl3的摩尔比为2.0:1.0;所述步骤2中的二氢吖啶类化合物和卤代芳烃化合物的摩尔比为1.0:2.0-1.0:5.0。
[0016]
优选的,所述步骤1中二氢吖啶类化合物在溶剂中的浓度为0.1-0.2摩尔/升。
[0017]
优选的,步骤1的具体制备过程为:将亚胺类化合物溶于装有溶剂的耐压管内,再加入alcl3,再用装有氩气的气球充闭耐压管,于30℃-200℃下加热搅拌反应2h-24h后,经过滤和减压浓缩后制备得到二氢吖啶类化合物。
[0018]
优选的,所述步骤1中的溶剂为乙醇和乙二醇中的一种或两种任意比例的混合物。
[0019]
优选的,步骤2的具体制备过程为:将二氢吖啶类化合物溶于装有甲苯的耐压管内,再依次缓慢加入溴苯和pd(oac)2后,用装有氩气的气球充闭耐压管,直至空气全部排空,封闭耐压管,于30℃-200℃下加热搅拌2h-24h后,经过滤和减压浓缩后,制备得到n-芳基吖啶类化合物。
[0020]
优选的,所述步骤3的具体制备过程为:将所述的n-芳基吖啶类化合物溶于装ch3cn的螺口瓶内,室温搅拌后缓慢加入氧化剂,再室温下搅拌反应2h-4h后,得到反应粗液a,将反应粗液a用dcm稀释并萃取,合并有机相,再经干燥剂干燥后得到反应粗液b,继续将反应粗液b用dcm稀释后加入圆底烧瓶中,再加入阴离子交换反应试剂,在室温下搅拌6-10h后,经减压浓缩和柱层析分离后得到吖啶四氟硼酸盐类光反应催化剂。
[0021]
优选的,所述阴离子交换反应试剂nabf4饱和水溶液;所述干燥剂采用na2so4,所述氧化剂采用二氯二氰基苯醌。
[0022]
与现有技术相比,本发明具有以下有益的技术效果:
[0023]
本发明提供的一种吖啶四氟硼酸盐类光催化剂及其合成方法,本发明首次提出以亚胺化合物为原料,在溶剂的存在下,单分子亚胺化合物自身缩合反应得到中间二氢吖啶经亚胺化合物、再经过渡金属催化偶联反应、氧化反应、阴离子交换反应后合成吖啶四氟硼酸盐类光反应催化剂,该方法操作简单,反应物稳定,反应周期短,能高效得到目标产物;
[0024]
进一步,相比于传统的通过苯炔前体、叠氮化物合成吖啶四氟硼酸盐的方法,反应原料合成简单,反应成本低,且操作安全;
[0025]
进一步,本发明的反应对底物的普适性较好,底物的来源广泛,在优化的反应条件下,目标产物易于分离,为光催化反应提供了更多光催化剂的选择。
附图说明
[0026]
图1为实施例1所制备的产物的1h nmr谱图;
[0027]
图2为实施例1所制备的产物的
13
c nmr谱图;
[0028]
图3为实施例2所制备的产物的1h nmr谱图;
[0029]
图4为实施例2所制备的产物的
13
c nmr谱图;
[0030]
图5为实施例3所制备的产物的1h nmr谱图;
[0031]
图6为实施例3所制备的产物的
13
c nmr谱图;
[0032]
图7为实施例4所制备的产物的1h nmr谱图;
[0033]
图8为实施例4所制备的产物的
13
c nmr谱图;
[0034]
图9为实施例5所制备的产物的1h nmr谱图;
[0035]
图10为实施例5所制备的产物的
13
c nmr谱图;
[0036]
图11为实施例6所制备的产物的1h nmr谱图;
[0037]
图12为实施例6所制备的产物的
13
c nmr谱图;
[0038]
图13为实施例7所制备的产物的1h nmr谱图;
[0039]
图14为实施例7所制备的产物的
13
c nmr谱图;
[0040]
图15为实施例8所制备的产物的1h nmr谱图;
[0041]
图16为实施例8所制备的产物的
13
c nmr谱图;
[0042]
图17为实施例9所制备的产物的1h nmr谱图;
[0043]
图18为实施例9所制备的产物的
13
c nmr谱图;
[0044]
图19为实施例10所制备的产物的1h nmr谱图;
[0045]
图20为实施例10所制备的产物的
13
c nmr谱图;
[0046]
图21为实施例11所制备的产物的1h nmr谱图;
[0047]
图22为实施例11所制备的产物的
13
c nmr谱图;
[0048]
图23为实施例12所制备的产物的1h nmr谱图;
[0049]
图24为实施例12所制备的产物的
13
c nmr谱图;
[0050]
图25为实施例13所制备的产物的1h nmr谱图;
[0051]
图26为实施例13所制备的产物的
13
c nmr谱图;
[0052]
图27为实施例14所制备的产物的1h nmr谱图;
[0053]
图28为实施例14所制备的产物的
13
c nmr谱图;
[0054]
图29为实施例15所制备的产物的1h nmr谱图;
[0055]
图30为实施例15所制备的产物的
13
c nmr谱图;
[0056]
图31为实施例16所制备的产物的1h nmr谱图;
[0057]
图32为实施例16所制备的产物的
13
c nmr谱图。
具体实施方式
[0058]
下面将结合实施例来详细说明本发明。需要说明的是,在不冲突的情况下,本技术
中的实施例及实施例中的特征可以相互组合。
[0059]
实施例1
[0060]
7,14-diphenyldibenzo[a,j]acridin-7-ium tetrafluoroborate的制备
[0061]
将0.4mmol的n-(萘-2-基)-1-苯基甲亚胺溶于装有1.5ml乙二醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌12h,反应完全后减压浓缩,再将浓缩所得固体溶于装有1.8ml甲苯的耐压管内,缓慢加入溴苯39.1mg、pd(oac)
2 3.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌4h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到76.8mg黄色固体化合物,产率为74%,所得产品结构式如下:
[0062][0063]
如图1和图2所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ8.26(d,j=9.3hz,2h),8.00(d,j=7.8hz,2h),7.94
–
7.82(m,4h),7.79
–
7.70(m,4h),7.68
–
7.57(m,4h),7.39
–
7.22(m,6h).
13
c nmr(100mhz,cdcl3):δ156.2,142.7,140.7,140.4,138.3,132.4,132.0,131.7,131.5,130.7,129.9,129.4,129.3,129.1,128.9,128.3,128.0,125.0,117.1.
[0064]
实施例2
[0065]
7-phenyl-14-(p-tolyl)dibenzo[a,j]acridin-7-ium tetrafluoroborate的制备
[0066]
将0.4mmol的n-(萘-2-基)-1-(对甲苯基)甲亚胺,溶于装有1.8ml乙醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌12h,反应完全后减压浓缩,再将浓缩所得固体溶于装有2.0ml甲苯的耐压管内,缓慢加入溴苯42.6mg、pd(oac)23.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌4h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到69.3mg黄色固体化合物,产率为65%,所得产品结构式如下:
[0067][0068]
如图3和图4所示,产品核磁表征:1h nmr(400mhz,dmso):δ8.56(d,j=9.4hz,2h),8.24(d,j=7.8hz,2h),8.05
–
7.98(m,3h),7.94
–
7.88(m,2h),7.78(t,j=7.9hz,2h),7.70(d,j=7.8hz,2h),7.49(d,j=7.8hz,2h),7.45
–
7.30(m,6h),2.68(s,3h).
13
c nmr(100mhz,dmso):δ155.9,143.0,141.3,140.5,139.0,138.1,132.6,132.3,132.0,131.9,130.5,129.4,129.3,129.2,129.2,128.8,128.8,128.3,124.8,117.9,21.8.
[0069]
实施例3
[0070]
14-(3,5-dimethylphenyl)-7-phenyldibenzo[a,j]acridin-7-iumtetrafluoroborate的制备
[0071]
将0.4mmol的1-(3,5-二甲基苯基)-n-(萘-2-基)甲亚胺,溶于装有1.5ml乙二醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌12h,反应完全后减压浓缩,再将浓缩所得固体溶于装有1.8ml甲苯的耐压管内,缓慢加入溴苯41.2mg、pd(oac)23.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌4h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到75.4mg黄色固体化合物,产率为69%,所得产品结构式如下:
[0072][0073]
如图5和图6所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ8.27(d,j=9.3hz,2h),8.01(d,j=7.8hz,2h),7.94
–
7.86(m,3h),7.71
–
7.61(m,4h),7.46(s,1h),7.42(d,j=8.7hz,2h),7.36
–
7.26(m,4h),7.16(s,2h),2.43(s,6h).
13
c nmr(100mhz,cdcl3):δ156.8,142.5,141.7,140.6,140.4,138.2,132.4,132.3,132.0,131.8,130.0,129.4,129.2,129.1,128.2,128.0,126.4,124.8,117.0,21.5.
[0074]
实施例4
[0075]
14-(4-methoxyphenyl)-7-phenyldibenzo[a,j]acridin-7-iumtetrafluoroborate的制备
[0076]
将0.4mmol的1-(4-甲氧基苯基)-n-(萘-2-基)甲亚胺,溶于装有1.6ml乙醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌12h,反应完全后减压浓缩,再将浓缩所得固体溶于装有
1.8ml甲苯的耐压管内,缓慢加入溴苯38.2mg、pd(oac)23.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌4h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到73.5mg黄色固体化合物,产率为67%,所得产品结构式如下:
[0077][0078]
如图7和图8所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ8.23(t,j=8.0hz,2h),8.06
–
7.81(m,7h),7.65(d,j=7.4hz,2h),7.53
–
7.24(m,10h),4.09(s,3h).
13
c nmr(100mhz,cdcl3):δ161.8,156.3,142.6,140.2,138.3,132.6,132.4,131.9,131.6,130.9,129.8,129.3,129.0,128.9,128.5,128.0,125.2,117.2,117.1,55.7.
[0079]
实施例5
[0080]
14-(4-(methylthio)phenyl)-7-phenyldibenzo[a,j]acridin-7-ium tetrafluoroborate的制备
[0081]
将0.4mmol的1-(4-(甲硫基)苯基)-n-(萘-2-基)甲亚胺,溶于装有2.0ml乙醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌12h,反应完全后减压浓缩,再将浓缩所得固体溶于装有2.4ml甲苯的耐压管内,缓慢加入溴苯43.6mg、pd(oac)23.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌4h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到73.4mg黄色固体化合物,产率为65%,所得产品结构式如下:
[0082][0083]
如图9和图10所示,产品核磁表征:1h nmr(400mhz,dmso):δ8.53(d,j=9.7hz,2h),8.23(d,j=7.5hz,2h),8.02
–
7.67(m,9h),7.50(d,j=7.3hz,2h),7.46
–
7.37(m,4h),7.33(d,j=9.3hz,2h),2.70(s,3h).
13
c nmr(100mhz,dmso):δ155.1,143.0,142.9,140.5,138.9,136.9,132.6,132.3,131.9,130.5,130.1,129.4,129.1,128.8,128.3,124.8,
117.8,15.1.
[0084]
实施例6
[0085]
14-(3,4-dimethylphenyl)-7-phenyldibenzo[a,j]acridin-7-iumtetrafluoroborate的制备
[0086]
将0.4mmol的1-(3,4-二甲基苯基)-n-(萘-2-基)甲亚胺,溶于装有1.6ml乙醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌12h,反应完全后减压浓缩,再将浓缩所得固体溶于装有1.8ml甲苯的耐压管内,缓慢加入溴苯40.6mg、pd(oac)
2 3.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌4h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到77.6mg黄色固体化合物,产率为71%,所得产品结构式如下:
[0087][0088]
如图11和图12所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ8.30(d,j=9.3hz,2h),8.04(d,j=7.9hz,2h),7.94(d,j=6.1hz,3h),7.74
–
7.65(m,4h),7.55(d,j=7.7hz,1h),7.45(d,j=8.7hz,2h),7.38
–
7.27(m,6h),2.62(s,3h),2.41(s,3h).
13
c nmr(100mhz,cdcl3):δ156.7,142.5,140.5,139.8,138.2,138.0,132.7,132.4,132.0,131.8,129.9,129.3,129.2,129.0,128.2,127.9,126.4,125.0,117.0,20.0,19.9.
[0089]
实施例7
[0090]
14-(3,4-dimethoxyphenyl)-7-phenyldibenzo[a,j]acridin-7-iumtetrafluoroborate的制备
[0091]
将0.4mmol的1-(3,4-二甲氧基苯基)-n-(萘-2-基)甲亚胺,装有2.0ml乙醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌12h,反应完全后减压浓缩,再将浓缩所得固体溶于装有1.8ml甲苯的耐压管内,缓慢加入溴苯41.8mg、pd(oac)23.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌4h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到74.1mg黄色固体化合物,产率为64%,所得产品结构式如下:
[0092][0093]
如图13和图14所示,产品核磁表征:1h nmr(400mhz,dmso):δ8.57(d,j=9.6hz,2h),8.26(d,j=7.9hz,2h),8.07
–
7.77(m,7h),7.53
–
7.42(m,5h),7.37(d,j=9.5hz,2h),7.19
–
7.08(m,2h),4.07(s,3h),3.63(s,3h).
13
c nmr(100mhz,dmso):δ155.8,152.1,151.6,142.9,140.5,138.9,132.9,132.5,132.3,132.0,130.4,129.4,129.3,128.8,128.3,125.0,122.1,117.7,115.1,113.1,56.6.
[0094]
实施例8
[0095]
14-(4-(tert-butyl)phenyl)-7-phenyldibenzo[a,j]acridin-7-iumtetrafluoroborate的制备
[0096]
将0.4mmol的1-(4-(叔丁基)苯基)-n-(萘-2-基)甲亚胺,溶于装有1.6ml乙二醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌12h,反应完全后减压浓缩,再将浓缩所得固体溶于装有2.0ml甲苯的耐压管内,缓慢加入溴苯39.8mg、pd(oac)
2 3.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌4h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到82.7mg黄色固体化合物,产率为72%,所得产品结构式如下:
[0097][0098]
如图15和图16所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ8.28(d,j=9.4hz,2h),8.02(d,j=7.9hz,2h),7.94
–
7.89(m,3h),7.80(d,j=8.1hz,2h),7.71
–
7.63(m,4h),7.51(d,j=8.1hz,2h),7.36(t,j=9.1hz,4h),7.24(t,j=8.2hz,2h),1.59(s,9h).
13
c nmr(100mhz,cdcl3):δ156.6,155.0,142.6,140.6,138.3,137.9,132.5,132.1,131.8,130.0,129.3,129.3,129.0,128.9,128.5,128.3,127.9,125.1,117.1,35.3,31.6.
[0099]
实施例9
[0100]
14-(naphthalen-2-yl)-7-phenyldibenzo[a,j]acridin-7-ium tetrafluoroborate的制备
[0101]
将0.4mmol的1-二(萘-2-基)甲亚胺,溶于装有1.5ml乙醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于
120℃下加热搅拌12h,反应完全后减压浓缩,再将浓缩所得固体溶于装有2.0ml甲苯的耐压管内,缓慢加入溴苯41.8mg、pd(oac)23.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌4h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到75.1mg黄色固体化合物,产率为66%,所得产品结构式如下:
[0102][0103]
如图31和图32所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ8.28(d,j=8.7hz,3h),8.15(d,j=8.3hz,1h),8.10(s,1h),8.00(d,j=7.9hz,2h),7.96
–
7.90(m,3h),7.87(d,j=8.2hz,1h),7.80
–
7.64(m,5h),7.57(t,j=7.5hz,2h),7.37(d,j=9.3hz,2h),7.29(d,j=8.8hz,2h),7.05(t,j=7.9hz,2h).
13
c nmr(100mhz,cdcl3):δ156.0,142.7,140.4,138.3,137.8,134.5,133.8,132.5,132.0,131.7,131.5,129.9,129.3,129.2,129.2,128.9,128.3,128.1,127.9,127.5,126.3,125.2,117.1.
[0104]
实施例10
[0105]
14-(4-nitrophenyl)-7-phenyldibenzo[a,j]acridin-7-ium tetrafluoroborate的制备
[0106]
将0.4mmol的n-(萘-2-基)-1-(4-硝基苯基)甲亚胺,溶于装有1.6ml乙醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于90℃下加热搅拌18h,反应完全后减压浓缩,再将浓缩所得固体溶于装有1.6ml甲苯的耐压管内,缓慢加入溴苯39.6mg、pd(oac)23.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于140℃下加热搅拌10h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到82.3mg黄色固体化合物,产率为73%,所得产品结构式如下:
[0107][0108]
如图19和图20所示,产品核磁表征:1h nmr(400mhz,dmso):δ8.72(d,j=8.2hz,
2h),8.61(d,j=9.4hz,2h),8.29(d,j=7.8hz,2h),8.10
–
8.02(m,3h),7.98(d,j=8.2hz,2h),7.95
–
7.88(m,2h),7.82(t,j=7.5hz,2h),7.48(t,j=8.0hz,2h),7.41(d,j=9.4hz,2h),7.23(d,j=8.7hz,2h).
13
c nmr(100mhz,dmso):δ152.9,149.3,147.2,143.1,140.8,138.8,132.7,132.4,132.0,131.7,130.7,129.7,129.2,128.7,128.5,127.0,124.4,117.9.
[0109]
实施例11
[0110]
14-(4-cyanophenyl)-7-phenyldibenzo[a,j]acridin-7-ium tetrafluoroborate的制备
[0111]
将0.4mmol的4-((萘-2-基亚氨基)甲基)苄腈,溶于装有2.2ml乙二醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于30℃下加热搅拌24h,反应完全后减压浓缩,再将浓缩所得固体溶于装有1.8ml甲苯的耐压管内,缓慢加入溴苯40.6mg、pd(oac)23.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于200℃下加热搅拌4h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到78.3mg黄色固体化合物,产率为72%,所得产品结构式如下:
[0112][0113]
如图21和图22所示,产品核磁表征:1h nmr(400mhz,cdcl3):δ8.21(d,j=9.4hz,2h),7.99(d,j=7.8hz,4h),7.92
–
7.83(m,5h),7.74(d,j=6.2hz,2h),7.65(t,j=7.4hz,2h),7.33
–
7.25(m,4h),7.21(d,j=8.7hz,2h).
13
c nmr(100mhz,cdcl3):δ153.2,145.4,142.9,140.2,138.3,134.6,132.6,131.9,131.5,131.2,130.1,129.3,129.0,128.6,128.4,127.9,124.7,118.1,117.2,114.2.
[0114]
实施例12
[0115]
3,7,11,14-tetraphenyldibenzo[a,j]acridin-7-ium tetrafluoroborate的制备
[0116]
将0.4mmol的1-苯基-n-(6-苯基萘-2-基)甲亚胺,溶于装有1.8ml乙醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于80℃下加热搅拌12h,反应完全后减压浓缩,再将浓缩所得固体溶于装有2.0ml甲苯的耐压管内,缓慢加入溴苯42.4mg、pd(oac)23.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于80℃下加热搅拌16h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水
溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到84.5mg黄色固体化合物,产率为63%,所得产品结构式如下:
[0117][0118]
如图23和图24所示,产品核磁表征:1h nmr(400mhz,dmso):δ8.67
–
8.56(m,4h),8.07
–
7.92(m,8h),7.88(d,j=7.6hz,4h),7.75
–
7.64(m,4h),7.55(t,j=7.5hz,4h),7.51
–
7.39(m,4h),7.36(d,j=9.1hz,2h).
13
c nmr(100mhz,dmso):δ155.4,142.9,140.8,140.8,140.4,139.0,138.3,133.3,132.4,132.2,132.0,131.5,129.6,129.5,129.0,128.7,128.1,127.8,127.5,126.4,124.8,118.3.
[0119]
实施例13
[0120]
7-(4-(methoxycarbonyl)phenyl)-14-(p-tolyl)dibenzo[a,j]acridin-7-ium tetrafluoroborate的制备
[0121]
将0.4mmol的n-(萘-2-基)-1-(对甲苯基)甲亚胺,溶于装有1.8ml乙醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于30℃下加热搅拌24h,反应完全后减压浓缩,再将浓缩所得固体溶于装有1.8ml甲苯的耐压管内,缓慢加入4-溴苯甲酸甲酯53.5mg、pd(oac)23.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于30℃下加热搅拌24h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到73.4mg黄色固体化合物,产率为65%,所得产品结构式如下:
[0122][0123]
如图25和图26所示,产品核磁表征:1h nmr(400mhz,dmso):δ8.53(d,j=8.8hz,4h),8.24(d,j=7.9hz,2h),8.06(d,j=8.0hz,2h),7.76(t,j=7.4hz,2h),7.68(d,j=7.8hz,2h),7.46(d,j=7.7hz,2h),7.43
–
7.32(m,6h),4.04(s,3h),2.66(s,3h).
13
c nmr(100mhz,dmso):δ165.8,156.2,142.7,142.6,141.3,140.7,138.0,133.1,132.7,132.6,130.6,129.6,129.4,129.3,129.2,128.8,128.3,124.8,117.8,53.3,21.7.
[0124]
实施例14
[0125]
7-(3-nitrophenyl)-14-(p-tolyl)dibenzo[a,j]acridin-7-ium tetrafluoroborate的制备
[0126]
将0.4mmol的n-(萘-2-基)-1-(对甲苯基)甲亚胺,溶于装有2.0ml乙醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于180℃下加热搅拌21h,反应完全后减压浓缩,再将浓缩所得固体溶于装有2.2ml甲苯的耐压管内,缓慢加入3-硝基溴苯50.1mg、pd(oac)23.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于140℃下加热搅拌6h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到85.5mg黄色固体化合物,产率为74%,所得产品结构式如下:
[0127][0128]
如图27和图28所示,产品核磁表征:1h nmr(400mhz,dmso):δ8.96(s,1h),8.84(d,j=8.4hz,1h),8.58(d,j=9.5hz,2h),8.40(d,j=8.0hz,1h),8.34
–
8.24(m,3h),7.80(t,j=7.3hz,2h),7.73(d,j=7.7hz,2h),7.50(d,j=8.6hz,4h),7.46
–
7.36(m,4h),2.69(s,3h).
13
c nmr(100mhz,dmso):δ156.3,150.2,143.1,141.4,140.8,139.5,138.0,135.4,133.5,132.7,132.7,130.6,129.4,129.3,129.3,128.8,128.4,127.1,124.9,124.8,118.0,21.8.
[0129]
实施例15
[0130]
7-(4-chlorophenyl)-14-(p-tolyl)dibenzo[a,j]acridin-7-ium tetrafluoroborate的制备
[0131]
将0.4mmol的n-(萘-2-基)-1-(对甲苯基)甲亚胺,溶于装有1.6ml乙醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于200℃下加热搅拌2h,反应完全后减压浓缩,再将浓缩所得固体溶于装有2.0ml甲苯的耐压管内,缓慢加入4-氯溴苯48.2mg、pd(oac)23.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于200℃下加热搅拌2h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌3h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm 20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到71.4mg黄色固体化合物,产率为63%,所得产品结构式如下:
[0132][0133]
如图29和图30所示,产品核磁表征:1h nmr(400mhz,dmso):δ8.57(d,j=9.4hz,2h),8.27(d,j=7.9hz,2h),8.10(d,j=8.3hz,2h),7.96(d,j=8.4hz,2h),7.78(t,j=7.5hz,2h),7.70(d,j=7.7hz,2h),7.5
–
7.34(m,8h),2.68(s,3h).
13
c nmr(100mhz,dmso):δ156.0,143.0,141.3,140.6,138.0,137.7,137.1,132.6,132.1,130.8,130.5,129.4,129.4,129.2,128.8,128.3,124.8,117.9,21.8.
[0134]
实施例16
[0135]
7,14-di-p-tolyldibenzo[a,j]acridin-7-ium tetrafluoroborate的制备
[0136]
将0.4mmol的n-(萘-2-基)-1-(对甲苯基)甲亚胺,溶于装有1.5ml乙二醇的耐压管内(配有磁力搅拌子),再加入26.6mg alcl3,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于120℃下加热搅拌12h,反应完全后减压浓缩,再将浓缩所得固体溶于装有2.2ml甲苯的耐压管内,缓慢加入4-甲基溴苯39.3mg、pd(oac)23.1mg,用气球充氩气至少3次,直至空气全部排空,封闭耐压管,于30℃下加热搅拌24h,反应完全后过滤,减压浓缩,将浓缩所得化合物溶于装有10ml ch3cn的螺口瓶内(配有磁力搅拌子),室温搅拌五分钟后缓慢加入氧化剂,室温搅拌6h,反应完成后将反应液用dcm(二氯甲烷)稀释并萃取,合并有机相,再经无水na2so4干燥后将反应粗液用dcm20ml稀释并加入50ml圆底烧瓶,后加入12ml nabf4饱和水溶液室温搅拌8h,将反应液减压浓缩,再经柱层析分离得到79.8mg黄色固体化合物,产率为73%,所得产品结构式如下:
[0137][0138]
如图31和图32所示,产品核磁表征:1h nmr(400mhz,dmso):δ8.56(d,j=9.4hz,2h),8.24(d,j=7.8hz,2h),7.85
–
7.74(m,6h),7.70(d,j=7.7hz,2h),7.49(d,j=7.7hz,2h),7.44
–
7.35(m,6h),2.68(s,6h).
13
c nmr(100mhz,dmso):δ155.8,143.1,142.3,141.2,140.4,138.1,136.5,132.6,132.6,132.3,130.5,129.4,129.2,128.8,128.5,128.2,124.8,117.9,21.8,21.5.
[0139]
由技术常识可知,本发明可以通过其他的不脱离其精神实质或必要特征的实施方案来实现。因此,上述公开的实施方案,就各方面而言,都只是举例说明,并不是仅有的。所有在本发明范围内或在等同于本发明的范围内的改变均被本发明包含。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。