1.本专利申请基于日本专利申请2020-072233号(于2020年4月14日提出申请)主张巴黎公约规定的优先权及利益,上述申请中记载的全部内容通过引用并入本说明书中。
2.本发明涉及核酸寡聚物的制造方法。本发明进一步详细地涉及包含硫代磷酸酯的核酸寡聚物的制造方法。
背景技术:
3.近年来,对核酸寡聚物在医疗领域的应用的兴趣不断高涨。例如可举出反义核酸、适配体、核酶及sirna等诱导rna干扰(rnai)的核酸等,它们被称作核酸药物。
4.核酸寡聚物可利用固相合成法合成,将在固相载体上延伸核酸而合成的核酸寡聚物从固相载体切出,接下来,例如对包含核糖的核酸寡聚物而言,对核糖的2’位的羟基的保护基进行脱保护并将其除去,从而制造作为目标的核酸寡聚物。具有硫代磷酸酯键的核酸寡聚物也作为有用的化合物而为人所知(专利文献1)。
5.现有技术文献
6.专利文献
7.专利文献1:国际公开第2017/068377号公报
技术实现要素:
8.发明所要解决的课题
9.具有硫代磷酸酯键的核酸寡聚物与仅包含磷酸二酯键的核酸寡聚物相比,有时稳定性成为问题。本发明的目的在于提供具有硫代磷酸酯键的核酸寡聚物的稳定化方法,以及包括将用该方法稳定化后的核酸寡聚物分离的工序的前述核酸寡聚物的高效的制造方法。
10.用于解决课题的手段
11.本技术发明人为了达成上述目的而反复进行了认真研究,结果果发现,使利用亚磷酰胺法生成的、具有硫代磷酸酯键的核酸寡聚物的粗产物经过反相柱层析,得到包含该核酸寡聚物的已纯化的洗脱级分,接下来,将与得到的洗脱级分接触的气氛设为一定浓度以下的非活性气体气氛,由此能够使具有该硫代磷酸酯键的核酸寡聚物稳定化。本发明提供具有硫代磷酸酯键的核酸寡聚物的稳定化方法,以及包括将用该方法稳定化后的核酸寡聚物分离的工序的、具有硫代磷酸酯键的核酸寡聚物的高效的制造方法。
12.本发明包括以下方式,但不限定于此。
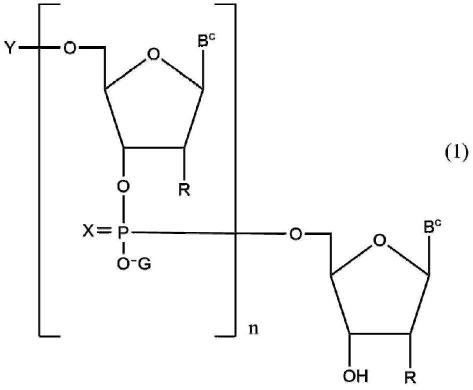
13.项1.核酸寡聚物的稳定化方法,其特征在于,将与包含式(1)所示的核酸寡聚物的溶液接触的气氛设为氧浓度10%以下的非活性气体气氛,前述溶液为从反相柱层析中洗脱的级分,
14.[化学式1]
[0015][0016]
式(1)中,
[0017]bc
各自独立地表示相同或彼此不同的核酸碱基,
[0018]
r相同或彼此不同,各自独立地表示氢原子、氟原子或oq基,
[0019]
q相同或彼此不同,各自独立地表示氢原子、甲基、2-甲氧基乙基、与核糖的4’位的碳原子键合的亚甲基、与核糖的4’位的碳原子键合的亚乙基、或与核糖的4’位的碳原子键合的乙叉基,
[0020]
x相同或彼此不同,各自独立地表示氧原子或硫原子,
[0021]
y表示氢原子或羟基的保护基,
[0022]
g表示铵离子、烷基铵离子、碱金属离子、氢离子或羟基烷基铵离子,
[0023]
n为满足式(2)的整数,并且,n个x中至多20%是硫原子,
[0024]
60≤n
ꢀꢀꢀꢀꢀꢀ
(2)。
[0025]
项2.经稳定化的纯化核酸寡聚物溶液的制造方法,其包括:
[0026]
使利用亚磷酰胺法生成的前述式(1)所示的核酸寡聚物的粗产物经过反相柱层析,得到包含式(1)的核酸寡聚物的已纯化的洗脱级分的工序;以及将与前项1中记载的前述洗脱级分接触的气氛设为氧浓度10%以下的非活性气体气氛的工序。
[0027]
项3.纯化核酸寡聚物的制造方法,其还包括:
[0028]
利用前项2中记载的工序制造纯化核酸寡聚物溶液的工序;和从前述纯化核酸寡聚物溶液中分离已纯化的核酸寡聚物的工序。
[0029]
项4.如前项1~3中任一项所述的方法,其中,氧浓度为5%以下。
[0030]
项5.如前项1~4中任一项所述的方法,其中,氧浓度为2.5%以下。
[0031]
项6.如前项2~5中任一项所述的方法,其中,前述反相柱层析为使用包含选自由单烷基铵盐及二烷基铵盐组成的组中的至少1种铵盐的流动相作为流动相的反相柱层析。
[0032]
项7.如前项2~6中任一项所述的方法,其中,前述反相柱层析为使用包含选自由二烷基铵盐组成的组中的至少1种铵盐的流动相作为流动相的反相柱层析。
[0033]
项8.如前项2~7中任一项所述的方法,其中,前述反相柱层析为使用包含选自由醇系水溶性有机溶剂及腈系水溶性有机溶剂组成的组中的至少1种水溶性有机溶剂的流动相作为流动相的反相柱层析。
[0034]
项9.如前项3所述的方法,其包括:将前述经稳定化的纯化核酸寡聚物溶液与具有
至少1个氧原子的c1-c4的有机溶剂进行混合,并分离所析出的核酸寡聚物。
[0035]
项10.如前项1~9中任一项所述的方法,其中,前述式(1)中,r各自独立地表示羟基或甲氧基。
[0036]
项11.如前项2~9中任一项所述的方法,其中,r为羟基。
[0037]
本发明还包含以下方式,但不限定于此。
[0038]
项1a.核酸寡聚物的制造方法,其包括:
[0039]
使利用亚磷酰胺法生成的式(1)所示的核酸寡聚物的粗产物经过反相柱层析,得到包含式(1)的核酸寡聚物的已纯化的洗脱级分的工序;以及
[0040]
将与前述洗脱级分接触的气氛设为氧浓度10%以下的非活性气体气氛,得到经稳定化的纯化核酸寡聚物溶液的工序,
[0041]
[化学式2]
[0042][0043]
式(1)中,
[0044]bc
各自独立地表示相同或彼此不同的核酸碱基,
[0045]
r相同或彼此不同,各自独立地表示氢原子、氟原子或oq基,
[0046]
q相同或彼此不同,各自独立地表示氢原子、甲基、2-甲氧基乙基、与核糖的4’位的碳原子键合的亚甲基、与核糖的4’位的碳原子键合的亚乙基、或与核糖的4’位的碳原子键合的乙叉基,
[0047]
x相同或彼此不同,各自独立地表示氧原子或硫原子,
[0048]
y表示氢原子或羟基的保护基,
[0049]
g表示铵离子、烷基铵离子、碱金属离子、氢离子或羟基烷基铵离子,
[0050]
n为满足式(2)的整数,并且,n个x中至多20%是硫原子,
[0051]
60≤n
ꢀꢀꢀꢀꢀꢀ
(2)。
[0052]
项2a.如项1a所述的核酸寡聚物的制造方法,其还包括:从前述稳定化的纯化核酸寡聚物溶液中分离已纯化的核酸寡聚物的工序。
[0053]
项3a.如项1a或项2a所述的核酸寡聚物的制造方法,其中,氧浓度为5%以下。
[0054]
项4a.如项1a或项2a所述的核酸寡聚物的制造方法,其中,氧浓度为2.5%以下。
[0055]
项5a.如前项1a~4a中任一项所述的制造方法,其中,前述反相柱层析的流动相为包含选自由单烷基铵盐及二烷基铵盐组成的组中的至少1种铵盐的流动相。
[0056]
项6a.如前项1a~4a中任一项所述的制造方法,其中,前述反相柱层析的流动相为包含选自由二烷基铵盐组成的组中的至少1种铵盐的流动相。
[0057]
项7a.如前项1a~6a中任一项所述的制造方法,其中,前述反相柱层析为使用包含选自由醇系水溶性有机溶剂及腈系水溶性有机溶剂组成的组中的至少1种水溶性有机溶剂的流动相作为流动相的反相柱层析。
[0058]
项8a.如项2a所述的制造方法,其包括:将前述经稳定化的纯化核酸寡聚物溶液与具有至少1个氧原子的c1-c4的有机溶剂进行混合,并分离所析出的核酸寡聚物。
[0059]
项9a.如前项1a~8a中任一项所述的制造方法,其中,前述式(1)中,r各自独立地表示羟基或甲氧基。
[0060]
项10a.如前项1a~8a中任一项所述的制造方法,其中,r为羟基。
[0061]
项11a.核酸寡聚物的稳定化方法,其特征在于,将与包含式(1)所示的核酸寡聚物的溶液接触的气氛设为氧浓度10%以下的非活性气体气氛,前述溶液为从反相柱层析中洗脱的级分,
[0062]
[化学式3]
[0063][0064]
式(1)中,
[0065]
各基团的定义与项[1a]中的定义相同。
[0066]
发明的效果
[0067]
通过本发明,具有硫代磷酸酯键的核酸寡聚物得以稳定化,可提供其高效的制造方法。
附图说明
[0068]
[图1]图1为示出利用亚磷酰胺法合成核酸寡聚物的例子的图。
具体实施方式
[0069]
针对核酸寡聚物的稳定化方法进行说明,其特征在于,将与包含前述式(1)所示的核酸寡聚物的溶液接触的气氛设为氧浓度10%以下的非活性气体气氛,前述溶液为从反相柱层析中洗脱的级分。
[0070]
前述式(1)中,bc所表示的核酸碱基(以下也记作“碱基”)可以是天然或非天然的
核酸碱基。作为该非天然的核酸碱基,可例示出天然或非天然的核酸碱基的修饰类似物。作为核酸碱基,典型地可例示出嘌呤化合物及嘧啶化合物,例如可例示出美国专利第3,687,808号、“concise encyclopedia of polymer science and engineering”,858~859页,kroschwitz j.i.编、john wiley&sons、1990及englisch等、angewandte chemie、international edition,1991,30卷,p.613中公开的核酸碱基。
[0071]
具体而言,例如可例示出腺嘌呤、异鸟嘌呤、黄嘌呤、次黄嘌呤及鸟嘌呤等嘌呤碱基;以及胞嘧啶、尿嘧啶及胸腺嘧啶等嘧啶碱基等。
[0072]
此外,作为bc所表示的核酸碱基,例如可例示出2-氨基腺嘌呤、2-氨基嘌呤、2,6-二氨基嘌呤等氨基衍生物;5-甲基尿嘧啶、5-甲基胞嘧啶、7-甲基鸟嘌呤、6-甲基嘌呤、2-丙基嘌呤等烷基衍生物;5-卤代尿嘧啶及5-卤代胞嘧啶;5-丙炔基尿嘧啶及5-丙炔基胞嘧啶;6-氮杂尿嘧啶、6-氮杂胞嘧啶及6-氮杂胸腺嘧啶;5-尿嘧啶(假尿嘧啶)、4-硫代尿嘧啶、5-(2-氨基丙基)尿嘧啶、5-氨基烯丙基尿嘧啶;8-卤化、氨基化、硫醇化、硫代烷基化、羟基化及其它的8-取代嘌呤;5-三氟甲基化及其它的5-取代嘧啶;6-氮杂嘧啶;n-2、n-6及o-6取代嘌呤(包括2-氨基丙基腺嘌呤);二氢尿嘧啶;3-脱氮-5-氮杂胞嘧啶;7-脱氮腺嘌呤;n6-甲基腺嘌呤、n6,n6-二甲基腺嘌呤;5-氨基-烯丙基-尿嘧啶;n3-甲基尿嘧啶;取代1,2,4-三唑;2-羟基吡啶;5-硝基吲哚;3-硝基吡咯;5-甲氧基尿嘧啶;尿嘧啶-5-氧基乙酸;5-甲氧基羰基甲基尿嘧啶;2-硫代尿嘧啶、5-甲基-2-硫代尿嘧啶;5-甲氧基羰基甲基-2-硫代尿嘧啶;5-甲基氨基甲基-2-硫代尿嘧啶;3-(3-氨基-3-羧基丙基)尿嘧啶;3-甲基胞嘧啶;n4-乙酰基胞嘧啶;2-硫代胞嘧啶;n6-甲基腺嘌呤;n6-异戊基腺嘌呤;2-甲硫基-n6-异戊烯基腺嘌呤;n-甲基鸟嘌呤;o-烷基化碱基等。
[0073]
当r表示oq基,q表示与核糖的4’位的碳原子键合的亚甲基、与核糖的4’位的碳原子键合的亚乙基或与核糖的4’位的碳原子键合的乙叉基时,该结构如下述式(3)中示出的lna-1、lna-2及lna-3的结构所示。
[0074]
[化学式4]
[0075][0076]
(式中,bc表示与前述相同的核酸碱基。)
[0077]
作为y所示的羟基的保护基,只要能够在酰胺法中作为保护基发挥功能,则能够没有特别制限地使用,例如,能够广泛地使用用于酰胺化合物的已知的保护基。y所示的羟基的保护基优选为以下基团。
[0078]
[化学式5]
[0079][0080]
(式中,r1、r2及r3相同或彼此不同,各自独立地表示氢或烷氧基。)
[0081]
作为前述烷氧基,例如可例示出甲氧基。
[0082]
式(1)的核酸寡聚物的链长例如可例示出n≥60、n≥80或n≥100的链长。作为链长的上限,例如可例示出n≤200。前述核酸寡聚物中,n个x中至多20%(包含20%)是硫原子。优选式(1)的核酸寡聚物具有1个以上的硫代磷酸酯键,进一步优选具有3个以上的硫代磷酸酯键。
[0083]
式(1)的核酸寡聚物也可以是例如dna或rna寡聚物、或者这些寡聚物中包含非天然型的核酸碱基而成的寡聚物。前述核酸寡聚物典型地为单链的dna或rna寡聚物。前述式(1)的核酸寡聚物中,取代基r优选各自独立地为羟基或甲氧基。本发明的方法适于作为式(1)的核酸寡聚物(取代基r各自独立地为羟基或甲氧基)的rna。进一步详细而言,适于制造包含取代基r为羟基的核苷酸和取代基r为甲氧基的核苷酸这两者的核酸寡聚物。
[0084]
<利用反相柱层析进行的核酸寡聚物的纯化方法>
[0085]
利用反相柱层析进行的分离通过下述方式实施:将包含烷基铵盐的流动相通液于包含填充剂的色谱柱,然后,将溶解有包含使用亚磷酰胺法合成的核酸寡聚物的反应产物的溶液通液于包含烷基铵盐的流动相,使前述核酸寡聚物吸附键合于色谱柱内,通过流动相中的有机溶剂浓度依次增大的梯度(gradient)使前述核酸寡聚物中包含的杂质、与作为目标的核酸分子分离并洗脱。
[0086]
对于利用反相柱层析得到的洗脱级分而言,在通常用于核酸的分离分析的层析的条件下以波长260nm的uv吸收对组成进行分析,并被选择和收集。从收集的级分中得到已纯化的目标物即具有规定量的硫代磷酸酯键的核酸寡聚物。作为前述分析法,例如能够使用非专利文献(handbook of analysis of oligonucleotides and related products,crc press)中记载的方法。
[0087]
关于前述反相柱层析的填充剂,作为成为疏水性的固定相的二氧化硅或聚合物,例如可例示出固定有选自苯基、碳原子数1~20的烷基及氰基丙基中的任1种以上的二氧化硅或聚合物。关于作为该填充剂的二氧化硅或聚合物,例如使用粒径为2μm以上、或5μm以上的二氧化硅或聚合物。
[0088]
反相柱层析中,进行洗脱并作为级分液而得到的是水溶性的流动相,作为成为流动相的溶剂系统,可例示出醇系有机溶剂(例如,甲醇、乙醇、2-丙醇或正丙醇)、腈系有机溶剂(例如,乙腈)及水。作为醇系有机溶剂,优选c1-c3醇,其中更优选甲醇。作为腈系有机溶剂,优选乙腈。各有机溶剂可以单独使用一种,也可以并用两种以上。
[0089]
前述反相柱层析的流动相通常使用包含烷基铵盐的流动相,洗脱液中也包含这些烷基铵盐。作为前述烷基铵盐,通常使用单烷基铵盐、二烷基铵盐及三烷基铵盐,优选使用单烷基铵盐及二烷基铵盐,更优选使用二烷基铵盐。形成单烷基铵盐的单烷基胺的碳原子
数优选为3~10,更优选为4~6,进一步优选为己基胺。形成二烷基铵盐的二烷基胺的碳原子数优选为4~10,更优选为5~9。优选的二烷基胺为二丁基胺。形成三烷基铵盐的三烷基胺的碳原子数优选为6~12,更优选为6~9,具体而言,可例示出三乙基胺。
[0090]
作为形成前述单烷基铵盐、二烷基铵盐及三烷基铵盐的酸,例如可例示出碳酸、乙酸、甲酸、三氟乙酸及丙酸。
[0091]
作为前述铵盐的浓度,通常为1~200mm,优选为5~150mm,更优选为20~100mm。
[0092]
流动相的ph范围通常为ph:6~8,优选为6.5~7.5。
[0093]
反相柱层析的温度通常为20~100℃,优选为30~80℃,更优选为40~70℃。
[0094]
利用反相柱层析得到的洗脱液级分中通常包含水、醇系有机溶剂、腈系有机溶剂、前述烷基铵盐及式(1)的核酸寡聚物。上述洗脱液中的水的量通常为90%~30%,优选为80%~40%,更优选为70%~40%。上述洗脱液中的醇系有机溶剂的量通常为0~20%,优选为0~15%,更优选为0%~10%。上述洗脱液中的腈系有机溶剂的量通常为10~70%,优选为20~60%,更优选为30~50%(以上的%均表示质量%)。
[0095]
上述洗脱级分中的烷基铵盐的摩尔浓度通常为1mm~200mm,优选为10mm~100mm。
[0096]
上述上述洗脱级分中的核酸寡聚物的浓度通常为0.05mg/ml~5mg/ml,优选为0.05mg/ml~1mg/ml,更优选为0.1mg/ml~0.5mg/ml。
[0097]
<利用反相色谱法得到的包含核酸寡聚物的洗脱级分的稳定化工序>
[0098]
氧浓度为10%以下的非活性气体气氛可以通过下述方式调节,例如,向包含核酸寡聚物的容器供给规定的氧浓度以下的非活性气体,测定并确认容器内的气氛中的氧浓度在前述设定浓度范围内。具体而言,能够通过下述方式调节:使容器内的气氛中流通氩或氮等高纯度的非活性气体、或将氧浓度配制成规定浓度的非活性气体;或者用前述非活性气体或调节浓度后的非活性气体置换容器内的气氛。
[0099]
氧浓度为10%以下的非活性气体气氛的配制可以在下述任一阶段进行:向容器供给前述洗脱级分前、或供给中、或向容器供给后。另外,也可以将它们组合来进行。
[0100]
上述非活性气体可举出氮气、氩气、氦气,但不限定于此。
[0101]
容器内的气氛置换方法可举出减压置换、加压置换、流动置换、基于鼓泡的置换、或基于冷冻脱气的置换,此时,可以施加超声波,也可以进行加热。更优选的方法为流动置换。
[0102]
前述非活性气体气氛下的氧浓度优选为10%以下,更优选为5.0%以下,进一步优选为2.5%以下,进一步更优选为0.1%以下。
[0103]
在前述非活性气体气氛下,通常可以将得到的洗脱级分于0℃~80℃、优选10℃~70℃、更优选20℃~60℃进行保存。
[0104]
利用反相色谱法得到的包含核酸寡聚物的洗脱级分例如也可以在保存之后施加选自用于分离核酸寡聚物的再沉淀工序、分液工序、超滤工序、脱保护工序及冷冻干燥工序这样的后处理工序中的单个或多个工序。
[0105]
再沉淀工序中,使经稳定化的溶液与不良溶剂接触,能够使核酸寡聚物析出并进行分离。如果需要,也可以由固液分离的状态除去液体部,然后,通过过滤等收集并分离所析出的核酸寡聚物。作为再沉淀工序的不良溶剂,可举出具有至少1个氧原子的c1-c4的有机溶剂(例如,c1-c4醇、四氢呋喃、二氧杂环己烷)。作为该溶剂,优选乙醇或异丙醇。
[0106]
分液工序中,在经稳定化的溶液中混合乙酸钠水溶液等酸性水溶液、水及食盐水等之中的至少一种,进一步加入不与水混和的有机溶剂,从而分液为水层和有机层,由此能够获取包含所期望的核酸寡聚物的水层。
[0107]
超滤工序中,能够使用超滤膜将在保存工序后的溶液中存在的核酸寡聚物与所期望的分子量以下的低分子成分分离。
[0108]
在核酸寡聚物的5’末端部位有保护基的情况下,为了对其进行脱保护,通过在保存工序后的溶液中混合乙酸水溶液等酸性水溶液、或将乙酸等酸性物质溶解于有机溶剂而成的溶液,能够对核酸寡聚物的保护基进行脱保护。
[0109]
冷冻干燥工序中,通过对冷冻的核酸寡聚物的水溶液进行减压而使水升华,从而能够将核酸寡聚物和水分分离。
[0110]
利用亚磷酰胺法进行的核酸寡聚物的合成能够依照已知的方法(例如,前述日本专利第5157168号公报或日本专利第5554881号公报中记载的方法)进行核酸延伸反应。针对利用亚磷酰胺法进行的核酸寡聚物的制造,可举出图1中示出的路线的rna的合成作为例子,参照以下示出的反应路径(缩合反应、氧化、脱保护)对核酸寡聚物的制造方法进行说明。
[0111]
表示反应路径的前述化学式中,ba表示可被保护的核酸碱基;tr表示保护基;x与前述定义相同,sp表示无机多孔载体的核苷结构以外的部分。
[0112]
具有核苷结构的无机多孔载体(sp-nu)及构成酰胺单体(am-1)的核苷的核酸碱基为与前述相同的核酸碱基或被保护基保护的核酸碱基。
[0113]
作为优选的酰胺单体(am-1)的例子,在下述化学式(am-1’)所表示的化合物中,r表示被保护的羟基时,作为具体的保护基,可例示出被叔丁基二甲基甲硅烷基(tbdms基)、双(2-乙酰氧基)甲基(ace基)、(三异丙基甲硅烷氧基)甲基(tom基)、(2-氰基乙氧基)乙基(cee基)、(2-氰基乙氧基)甲基(cem基)、对甲苯磺酰基乙氧基甲基(tem基)、(2-氰基乙氧基)甲氧基甲基(emm基)等保护的、tbdms酰胺(tbdms rna amidites,商品名,chemgenes corporation)、ace酰胺、tom酰胺、cee酰胺、cem酰胺、tem酰胺(chakhmakhcheva的总论:protective groups in the chemical synthesis of oligoribonucleotides、rus sian journal of bioorganic chemistry,2013,vol.39,no.1,pp.1-21.)、emm酰胺(国际公开第2013/027843号中记载)等。
[0114]
[化学式6]
[0115][0116]
(式中,r表示与前述相同的基团,ba表示可被保护的核酸碱基。)
[0117]
[rna的固相合成]
[0118]
对无机多孔载体(sp-nu)的tr基进行脱保护得到固相载体(am-2)。然后,使酰胺单体(am-1)和固相载体(am-2)进行缩合反应,得到反应产物(am-3)。然后,对反应产物(am-3)进行氧化,得到产物(am-4)。然后,对产物(am-4)进行脱保护(-tr),得到产物(am-5)。接下来,使酰胺单体(am-1)和产物(am-5)进一步进行缩合反应,从而延伸磷酸二酯键。如此,以成为所期望的序列的方式对延伸的寡核苷酸链末端的5’位的羟基重复进行必要的一系列的脱保护、缩合反应、氧化的循环,然后,从固相载体切出,由此能够制造所期望的序列的核酸分子。该合成也可以使用采用亚磷酰胺法的核酸自动合成装置等进行。此处以rna为例进行说明,但也能够适用于包含核糖核苷酸之外的核苷酸的核酸化合物。
[0119]
对tr基进行脱保护的工序中,对担载于固相载体上的rna链末端的5’位的羟基的保护基进行脱保护。作为保护基,使用三苯甲基系保护基(典型而言,dmtr基)。脱保护能够使用酸来进行。作为脱保护用的酸,例如可举出三氟乙酸、三氯乙酸、二氯乙酸、三氟甲磺酸、甲磺酸、盐酸、乙酸、对甲苯磺酸等。
[0120]
缩合工序中,使利用前述脱保护工序进行了脱保护的rna链末端的5’位的羟基与核苷亚磷酰胺键合,生成亚磷酸酯。作为前述核苷亚磷酰胺,使用5’位的羟基被保护基(例如dmtr基)保护的核苷亚磷酰胺。
[0121]
另外,缩合工序能够使用激活前述核苷亚磷酰胺的激活剂来进行。作为激活剂,例如可举出5-苄硫基-1h-四氮唑(btt)、1h-四氮唑、4,5-二氰基咪唑(dci)、5-乙硫基-1h-四氮唑(ett)、n-甲基苯并咪唑鎓三氟甲磺酸盐(n-mebit)、苯并咪唑鎓三氟甲磺酸盐(bit)、n-苯基咪唑鎓三氟甲磺酸盐(n-phimt)、咪唑鎓三氟甲磺酸盐(imt)、5-硝基苯并咪唑鎓三氟甲磺酸盐(nbt)、1-羟基苯并三唑(hobt)及5-(双-3,5-三氟甲基苯基)-1h-四氮唑(activator-42)等。
[0122]
在缩合工序之后,也可以适当对未反应的5’位的羟基进行加帽。加帽能够使用乙酸酐-四氢呋喃溶液、苯氧乙酸酐/n-甲基咪唑溶液等已知的加帽溶液来进行。
[0123]
氧化工序是对利用前述缩合工序形成的亚磷酸酯进行氧化的工序。氧化工序能够使用氧化剂进行。作为氧化剂,可举出碘、间氯过氧苯甲酸、叔丁基过氧化氢、2-过氧化丁酮、双(三甲基甲硅烷基)过氧化物、1,1-二氢过氧环十二烷(1,1-dihydroperoxycyclododecane)及过氧化氢等。
[0124]
在将亚磷酸三酯基转化成硫代磷酸三酯基的情况下,作为“氧化剂”,例如能够使用硫、3h-1,2-苯并二硫醇-3-酮-1,1-二氧化物(beaucage试剂)、3-氨基-1,2,4-二噻唑-5-硫酮(adtt)、5-苯基-3h-1,2,4-二噻唑-3-酮(pos)、[(n,n-二甲基氨基亚甲基)氨基]-3h-1,2,4-二噻唑啉-3-硫酮(ddtt)及苯乙酰二硫化物(pads)。该氧化剂能够以成为0.001~2m的浓度的方式用适宜的溶剂稀释来使用。作为反应中使用的溶剂,只要不参与反应,则没有特别限定,例如可举出二氯甲烷、乙腈、吡啶或它们的任意比例的混合溶剂。
[0125]
氧化工序可以在前述加帽操作之后进行,反之,也可以在氧化工序之后进行加帽操作,该顺序没有限定。
[0126]
通过在氧化工序后回到脱保护工序并根据应合成的核酸寡聚物的核苷酸序列重复上述的缩合反应、氧化、脱保护这一系列的工序,能够合成具有所期望的序列的rna。
[0127]
在具有所期望的序列的核酸寡聚物的合成结束后,使用氨或胺化合物,从固相载体切断并回收rna链。
[0128]
作为此处的胺化合物,例如可举出甲基胺、乙基胺、异丙基胺、亚乙基二胺、二乙基胺及三乙基胺等。
[0129]
这样得到的核酸寡聚物的链长例如可例示出n≥60、n≥80或n≥100、并且n≤200的链长。
[0130]
对磷酸保护基进行脱保护的工序在具有所期望的序列的核酸的合成结束后使胺化合物发挥作用以对磷酸部分的保护基进行脱保护。作为胺化合物,例如可举出记载的二乙基胺等。
[0131]
在有核糖的2’位或3’位的羟基的保护基的情况下,能够依照国际公开第2006/022323号公报)、国际公开第2013/027843号公报、或国际公开第2019/208571号公报中记载的方法来除去。
[0132]
实施例
[0133]
以下,利用实施例进一步详细地说明本发明,但本发明不限定于此。
[0134]
测定方法
[0135]
以下的试验中使用的各测定方法如下所示。
[0136]
(测定方法1:rna的纯度的测定方法)
[0137]
用反相色谱法洗脱的级分溶液中的rna的纯度的测定利用hplc进行。利用hplc(波长260nm,色谱柱dnapactm pa200、4.0mm
×
250mm、8.0μm)将分取的rna分离为各成分,由得到的色谱图的总面积值中的主产物的面积值计算rna的纯度。将hplc测定条件示于下述表1。
[0138]
[表1]
[0139][0140]
(测定方法2:氧浓度的测定)
[0141]
与用反相色谱法洗脱的级分溶液接触的气氛的氧浓度使用iijima electronics corp.制的pack keeper(残氧测定仪)(residual oxygen meter)测定。在氧浓度测定前,通过测定空气中及纯氮中氧浓度来校准装置后,将装置中附带的针插入用隔垫等盖着的烧瓶等容器,测定体系中气相部分的氧浓度。实时显示氧浓度的测定值,将测定值稳定的值作为其气氛的氧浓度。
[0142]
[参考例1]
[0143]
rna的基于酰胺法的固相合成
[0144]
合成具有以下示出的i的核酸序列的rna。该链由103个碱基长度组成。
[0145]
链i:a*u*a*acucaauuuguaaaaaaguuuuagagcuagaaauagcaaguuaaaauaaggcuaguccguuaucaacuugaaaaaguggcaccgagucggugcuuuu*u*u*u(5
’‑
3')(序列号1)
[0146]
前述序列的标注中,序列的说明中,u表示尿苷、c表示胞苷、a表示腺苷、或g表示鸟苷。核苷酸之间的标记*表示连接核苷酸的磷酸键为硫代磷酸酯。
[0147]
该rna是基于亚磷酰胺法使用核酸合成仪(akta oligopi lot plus100 ge healthcare公司)从3’侧向5’侧进行合成的。合成以63μmol规模实施。另外,使用下述试剂进行该合成:作为rna酰胺,分别使用下述式的尿苷emm酰胺(国际公开第2013/027843号的实施例2中记载)、胞苷emm酰胺(国际公开第2013/027843号的实施例3中记载)、腺苷emm酰胺(国际公开第2013/027843号的实施例4中记载)及鸟苷emm酰胺(国际公开第2013/027843号的实施例5中记载),使用多孔玻璃作为固相载体,使用二氯乙酸甲苯溶液作为脱保护溶液,使用5-苄硫基-1h-四氮唑作为缩合剂,使用碘溶液作为氧化剂,使用3-氨基-1,2,4-二噻唑-5-硫酮作为硫化剂,使用苯氧基乙酸酐溶液和n-甲基咪唑溶液作为加帽溶液。核酸延伸结束后,通过使二乙基胺溶液作用于载体上的核酸从而选择性地对磷酸部分的氰基乙基保护基进行脱保护。此处,emm为(2-氰基乙氧基)甲氧基甲基的缩写。
[0148]
[化学式7]
[0149][0150]
从固相合成后的固相载体的切出和脱保护依照国际公开第2013/027843号中记载的方法。即,加入氨水溶液和乙醇,静置一段时间后,过滤固相载体,蒸馏除去溶剂。然后,使用四丁基氟化铵进行羟基的脱保护。使用注射用蒸馏水以成为所期望的浓度的方式溶解得到的rna。
[0151]
rna的分取纯化
[0152]
在下述表2的条件下进行合成的rna的柱层析纯化。其中,在纯化前,在色谱柱内将流动相a以流速4.7ml/min通液12.5分钟后添加样本。在保留时间94.2分钟-95.8分钟为止进行分取,利用hplc对得到的溶液进行分析。需要说明的是,利用前述测定方法1中记载的
方法计算纯度。其结果是,分取的级分中的rna的纯度为94%。
[0153]
[表2]
[0154][0155]
分取溶液的保存稳定性的确认
[0156]
[实施例1]
[0157]
将前述参考例1中分取纯化的包含rna的级分的溶液0.5ml放入容量10ml的玻璃制顶空进样瓶(gl sciences公司),用隔垫及螺旋盖(crimp cap)(gl sciences公司)密封。进一步在密封的体系内,将从氮气瓶吹出氮的针、用于将吹出的氮抽出的针以及氧气测定仪的测定用针插入隔垫,通过使氮流动来将体系内气氛置换为氮,使气氛中的氧浓度为0.0%。此处,使用前述测定方法2中记载的方法测定氧浓度。然后,将放入有分取溶液的玻璃制顶空进样瓶放入调温至60℃的恒温箱(kenis公司),静置8小时。静置后,将从恒温箱取出的顶空进样瓶冷却至室温,利用前述测定方法1中记载的方法计算rna的纯度时,为83%。
[0158]
[实施例2]
[0159]
将前述参考例1中分取纯化的包含rna的级分的溶液0.5ml放入容量10ml的玻璃制顶空进样瓶(gl sciences公司),用隔垫及螺旋盖(gl sciences公司)密封。进一步在密封的体系内,将从氮气瓶吹出氮的针、用于将吹出的氮抽出的针以及氧气测定仪的测定用针插入隔垫,通过使氮流动来将体系内气氛置换为氮,使气氛中的氧浓度为2.1%。此处,使用前述测定方法2中记载的方法测定氧浓度。然后,将放入有分取溶液的玻璃制顶空进样瓶放入调温至60℃的恒温箱(kenis公司),静置8小时。静置后,将从恒温箱取出的顶空进样瓶冷却至室温,利用前述测定方法1中记载的方法计算rna的纯度时,为77%。
[0160]
[实施例3]
[0161]
将前述参考例1中分取纯化的包含rna的级分的溶液0.5ml放入容量10ml的玻璃制顶空进样瓶(gl sciences公司),用隔垫及螺旋盖(gl sciences公司)密封。进一步在密封的体系内,将从氮气瓶吹出氮的针、用于将吹出的氮抽出的针以及氧气测定仪的测定用针插入隔垫,通过使氮流动来将体系内气氛置换为氮,使气氛中的氧浓度为7.3%。此处,使用前述测定方法2中记载的方法测定氧浓度。然后,将放入有分取溶液的玻璃制顶空进样瓶放
入调温至60℃的恒温箱(kenis公司),静置8小时。静置后,将从恒温箱取出的顶空进样瓶冷却至室温,利用前述测定方法1中记载的方法计算rna的纯度时,为73%。
[0162]
[比较例1]
[0163]
将前述参考例1中分取纯化的包含rna的级分的溶液0.5ml放入容量10ml的玻璃制顶空进样瓶(gl sciences公司),不对气氛进行氮置换,用隔垫及螺旋盖(gl sciences公司)密封。然后,将放入有分取溶液的玻璃制顶空进样瓶放入调温至60℃的恒温箱(kenis公司),静置8小时。静置后,将从恒温箱取出的顶空进样瓶冷却至室温后,利用前述测定方法1中记载的方法计算级分中的rna的纯度时,为60%。
[0164]
[表3]
[0165] o2浓度[%]hplc测定纯度
1)
核酸的保留率
2)
实施例1083%88%实施例22.177%82%实施例37.373%77%比较例120.960%64%
[0166]
1)将参考例1中分取纯化的核酸(rna)于60℃、在规定的氧浓度条件下静置保存8小时后,利用hplc测定的核酸的纯度(%)
[0167]
2)核酸的保留率(%)=保存后的核酸的hplc纯度/参考例2中分取纯化时的核酸的纯度
[0168]
从保存的分取溶液回收rna
[0169]
[实施例4]
[0170]
将实施例1中气氛的氧浓度设为0.0%并静置8小时后的溶液0.4ml放入容量15ml的聚丙烯制离心管(corning公司),追加乙酸钠水溶液(3m,ph=5.2)0.2ml、乙醇1.2ml。以10分钟、3000g、25℃的条件对得到的浆状溶液进行离心,除去上清液。然后,将放入70%乙醇水溶液1ml,以10分钟、3000g、25℃的条件进行离心并除去上清液的操作重复2次,得到作为沉淀的rna。将得到的rna溶解于水0.4ml,利用前述测定方法1中记载的方法计算级分中的rna的纯度时,纯度为77%。
[0171]
[比较例2]
[0172]
将比较例1中静置8小时后的分取溶液0.4ml放入容量15ml的聚丙烯制离心管(corning公司),追加乙酸钠水溶液(3m,ph=5.2)0.2ml、乙醇1.2ml。以10分钟、3000g、25℃的条件对得到的浆状溶液进行离心,除去上清液。然后,将放入70%乙醇水溶液1ml,以10分钟、3000g、25℃的条件进行离心并除去上清液的操作重复2次,得到rna。将得到的rna溶解于水0.4ml,利用前述测定方法1中记载的方法计算级分中的rna的纯度时,纯度为64%。
[0173]
[参考例2]
[0174]
rna的基于酰胺法的固相合成
[0175]
合成具有以下示出的ii的核酸序列的rna。该链由67个碱基长度组成。
[0176]
链ii:am*gm*cm*amumamgmcaaguuamaaauaaggmc*u*amg*u*c*cmguuaucaamcmumumgmamamamamamgmumggcacmcmgmagucggmumgmcm*um*um*u(5
’‑
3')(序列号2)
[0177]
前述序列的标注中,核苷酸之间的标记*表示连接核苷酸的磷酸键为硫代磷酸酯。字母am,um,cm,gm表示2’羟基被替换成甲氧基的核苷酸。该rna是基于亚磷酰胺法使用
核酸合成仪(akta oligopi lot plus100 ge healthcare公司)从3’侧向5’侧进行合成的。合成以53μmol规模实施。另外,使用下述试剂进行该合成:作为rna酰胺,使用尿苷emm酰胺(国际公开第2013/027843号的实施例2中记载)、胞苷emm酰胺(国际公开第2013/027843号的实施例3中记载)、腺苷emm酰胺(国际公开第2013/027843号的实施例4中记载)及鸟苷emm酰胺(国际公开第2013/027843号的实施例5中记载);和下述式的尿苷2’ome酰胺、胞苷2’ome酰胺、腺苷2’ome酰胺及鸟苷2’ome酰胺,使用多孔玻璃作为固相载体,使用二氯乙酸甲苯溶液作为脱保护溶液,使用5-苄硫基-1h-四氮唑作为缩合剂,使用碘溶液作为氧化剂,使用3-氨基-1,2,4-二噻唑-5-硫酮作为硫化剂,使用苯氧基乙酸酐溶液和n-甲基咪唑溶液作为加帽溶液。核酸延伸结束后,通过使二乙基胺溶液作用于载体上的核酸从而选择性地对磷酸部分的氰基乙基保护基进行脱保护。此处,emm为(2-氰基乙氧基)甲氧基甲基的缩写。
[0178]
[化学式8]
[0179][0180]
从固相合成后的固相载体的切出和脱保护依照国际公开第2013/027843号中记载的方法。即,加入氨水溶液和乙醇,静置一段时间后,过滤固相载体,蒸馏除去溶剂。然后,使用四丁基氟化铵进行羟基的脱保护。使用注射用蒸馏水以成为所期望的浓度的方式溶解得到的rna。
[0181]
rna的分取纯化
[0182]
在下述表4的条件下进行柱层析纯化。其中,在纯化前,在色谱柱内将流动相a以流速4.7ml/min通液12.5分钟后添加样本。在保留时间66.7分钟-70.9分钟为止进行分取,利用hplc对得到的溶液进行分析。需要说明的是,利用前述测定方法1中记载的方法计算纯度。其结果是,纯度为94%。使用该分取纯化的rna溶液进行以下实施例及比较例的实验。
[0183]
[表4]
[0184][0185]
分取溶液的保存稳定性的确认
[0186]
[实施例5]
[0187]
将前述参考例2中分取纯化的包含rna的级分的溶液0.5ml放入容量10ml的玻璃制顶空进样瓶(gl sciences公司),用隔垫及螺旋盖(gl sciences公司)密封。进一步在密封的体系内,将从氮气瓶吹出氮的针、用于将吹出的氮抽出的针以及氧气测定仪的测定用针插入隔垫,通过使氮流动来将体系内气氛置换为氮,使气氛中的氧浓度为0.0%。此处,使用前述测定方法2中记载的方法测定氧浓度。然后,将放入有分取溶液的玻璃制顶空进样瓶放入调温至60℃的恒温箱(kenis公司),静置8小时。静置后,将从恒温箱取出的顶空进样瓶冷却至室温,利用前述测定方法1中记载的方法计算rna的纯度时,为87%。
[0188]
[实施例6]
[0189]
将前述参考例2中分取纯化的包含rna的级分的溶液0.5ml放入容量10ml的玻璃制顶空进样瓶(gl sciences公司),用隔垫及螺旋盖(gl sciences公司)密封。进一步在密封的体系内,将从氮气瓶吹出氮的针、用于将吹出的氮抽出的针以及氧气测定仪的测定用针插入隔垫,通过使氮流动来将体系内气氛置换为氮,使气氛中的氧浓度为2.1%。此处,使用前述测定方法2中记载的方法测定氧浓度。然后,将放入有分取溶液的玻璃制顶空进样瓶放入调温至60℃的恒温箱(kenis公司),静置8小时。静置后,将从恒温箱取出的顶空进样瓶冷却至室温,利用前述测定方法1中记载的方法计算rna的纯度时,为84%。
[0190]
[实施例7]
[0191]
将前述参考例2中分取纯化的包含rna的级分的溶液0.5ml放入容量10ml的玻璃制顶空进样瓶(gl sciences公司),用隔垫及螺旋盖(gl sciences公司)密封。进一步,在密封的体系内,将从氮气瓶吹出氮的针、用于将吹出的氮抽出的针以及氧气测定仪的测定用针插入隔垫,通过使氮流动来将体系内气氛置换为氮,使气氛中的氧浓度为4.5%。此处,使用前述测定方法2中记载的方法测定氧浓度。然后,将放入有分取溶液的玻璃制顶空进样瓶放入调温至60℃的恒温箱(kenis公司),静置8小时。静置后,将从恒温箱取出的顶空进样瓶冷却至室温,利用前述测定方法1中记载的方法计算rna的纯度时,为83%。
[0192]
[比较例3]
[0193]
将前述参考例2中分取纯化的包含rna的级分的溶液0.5ml放入容量10ml的玻璃制顶空进样瓶(gl sciences公司),不对气氛进行氮置换,用隔垫及螺旋盖(gl sciences公司)密封。然后,将放入有分取溶液的玻璃制顶空进样瓶放入调温至60℃的恒温箱(kenis公
司),静置8小时。静置后,将从恒温箱取出的顶空进样瓶冷却至室温后,利用前述测定方法1中记载的方法计算级分中的rna的纯度时,纯度为79%。
[0194]
[表5]
[0195] o2浓度[%]hplc测定纯度
1)
核酸的保留率
2)
实施例5087%92%实施例62.184%89%实施例74.583%88%比较例320.979%84%
[0196]
1)将参考例2中分取纯化的核酸(rna)于60℃、在规定的氧浓度条件下静置保存8小时后,利用hplc测定的核酸的纯度(%)
[0197]
2)核酸的保留率(%)=保存后的核酸的hplc纯度/参考例2中分取纯化时的核酸的纯度
[0198]
产业上的可利用性
[0199]
能够使反相色谱法的洗脱级分中的具有硫代磷酸酯键的核酸寡聚物稳定化,能够高效地制造已纯化的具有硫代磷酸酯键的核酸寡聚物(例如,rna)。
[0200]
[序列表自由文本]
[0201]
序列表的序列号1及2表示依照本发明的制造方法制造的寡核苷酸的碱基序列。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。