1.本发明涉及药物制剂技术领域,具体涉及一种克拉霉素片及其制备方法。
背景技术:
2.克拉霉素片为大环内酯类抗生素,克拉霉素是红霉素的衍生物,其可透过细菌的细胞壁进而与细菌核糖体50s亚基可逆行的结合而产生阻滞转肽及以为作用,能有效抑制链的延长,减少rna依耐性蛋白质的合成,发挥抑菌作用,其抑菌活性是红霉素的两倍以上。与其他传统大环内酯类抗生素比较,克拉霉素的药效学及药物代谢动力学性质均有明显改善,口服在胃肠道吸收迅速,绝对生物利用度可达55%。
3.克拉霉素的临床剂型多为口服制剂,包括片剂、胶囊、糖浆剂、混悬剂。其中,克拉霉素片是首批289个需要在2018年前完成仿制药一致性评价的品种之一。如何才能做到跟原研药一致的体内生物等效性?首先需要达到的就是体外的一致性,即两者在各种具有区分力的溶出介质中溶出曲线相似。承接克拉霉素片一致性评价研究工作的四川省药检所的前期研究表明,目前市场上销售的克拉霉素片与原研药体外溶出曲线在ph6.8磷酸盐缓冲液介质中区别明显,而该介质为最有区分力的介质。因此,十分有必要研发一种与原研药体外溶出曲线相似的克拉霉素片的制备方法,才能完成克拉霉素片的一致性评价。
技术实现要素:
4.为了克服现有技术的不足,本发明的目的在于提供一种克拉霉素片的制备方法,该方法重现性好、能够充分保证产品批间质量的一致性,由该方法制得的克拉霉素片与原研药在多种溶出介质中的溶出曲线均相似,其中在最有区分力的ph6.8磷酸盐缓冲液介质中相似度可大于72。
5.为解决上述问题,本发明所采用的技术方案如下:
6.一种克拉霉素片的制备方法,其包括以下步骤:
7.s1、原辅料预处理:将克拉霉素原料、预胶化淀粉、微晶纤维素、交联羧甲基纤维素钠和羟丙纤维素分别过筛;
8.s2、配制增溶剂和粘合剂:向司盘80溶液中加入95%乙醇,搅拌均匀,配制成浓度为10%的司盘-80醇溶液;将聚维酮k30加入纯化水中,搅拌溶解,制得浓度为8%的聚维酮k30粘合剂;
9.s3、制粒:将处方量的预胶化淀粉、微晶纤维素和交联羧甲基纤维素钠投入湿法制粒机中,搅拌均匀,加入司盘-80醇溶液,继续搅拌均匀;加入处方量的克拉霉素原料和羟丙纤维素,搅拌均匀后加入聚维酮k30粘合剂继续搅拌,制粒、湿整粒后干燥;
10.s4、整粒、总混:向干燥后的颗粒加入处方量的二氧化硅、硬脂酸和硬脂酸镁并进行干整粒,整粒后的颗粒转移到混合机中进行总混,得到总混颗粒;
11.s5、压片、包衣:将总混颗粒投料至压片机进行压片,得到素片;对素片进行薄膜包衣,得到克拉霉素片。
12.作为本发明优选的实施方式,所述步骤s3中干燥的进风温度为70~80℃、进风风量为500~1500m3/h;当物料温度达到42~47℃、干燥失重在2.0~3.5%时,即可出料。
13.作为本发明优选的实施方式,所述步骤s5中包衣的片床温度为37~45℃。
14.作为本发明优选的实施方式,所述克拉霉素片由以下按质量百分数计的各组分组成:克拉霉素48.9%、微晶纤维素20.1%、羟丙纤维素6.0%、预胶化淀粉15.1%、交联羧甲基纤维素钠5.0%、司盘-80 0.1%、聚维酮k30 2.6%、二氧化硅0.3%、硬脂酸1.0%、硬脂酸镁0.8%。
15.作为本发明优选的实施方式,所述步骤s1的原辅料分别过40-60目筛。
16.作为本发明优选的实施方式,所述步骤s3中加入预胶化淀粉、微晶纤维素和交联羧甲基纤维素钠后的混合时间为2min、搅拌桨转速为180~270rpm,切割刀转速为1500-2000rpm;加入10%司盘-80醇溶液后的搅拌桨转速为50~100rpm,切割刀转速为1500-2000rpm;加入克拉霉素原料和羟丙纤维素后的搅拌桨转速为180~270rpm、切割刀转速为1500-2000rpm、混合时间为3min;制粒的时间为6~15分钟、转速为800-1500rpm。
17.作为本发明优选的实施方式,所述步骤s3中每1万片批量加65-70g司盘-80醇溶液,每1万片批量加入1600-1700g聚维酮k30粘合剂。
18.作为本发明优选的实施方式,所述步骤s4中总混的混合时间为5-10min、转速为10rpm。
19.作为本发明优选的实施方式,所述总混颗粒的干燥失重为1.5~4.0%。
20.作为本发明优选的实施方式,所述步骤s5中压片的参数为:硬度为9.0~15.0kg、片厚为5.3~5.9mm、片中差异为预压片重的
±
4.5%。
21.本发明的目的之二在于提供一种由上述的制备方法所制得的克拉霉素片。
22.相比现有技术,本发明的有益效果在于:
23.通过本发明的制备方法所制得的样品与克拉霉素片的原研药相比,两者在水、ph1.2盐酸溶液、ph5.0醋酸盐缓冲液、ph6.0磷酸氢二钠-枸橼酸缓冲液和ph6.8磷酸盐缓冲液这五种溶出介质中的溶出曲线均相似,且最有区分力的ph6.8磷酸盐缓冲液介质中相似度可大于72。在有关物质检测结果方面,本发明制备的克拉霉素片可略优于原研药;在样品的稳定性方面,将样品置于60
±
2℃、rh75%条件下加速考察1个月和40
±
2℃、rh75%条件下加速考察3个月后的溶出曲线与原研药相似,有关物质无明显增长趋势。可见,按照本发明制备的样品稳定性较好,能够保证产品在效期内质量稳定,安全有效。
24.综上,本发明所提供的克拉霉素片的制备方法通过控制司盘-80醇溶液的加入方式等使所制得的克拉霉素片与原研药具有较好的溶出曲线,处方设计合理,制备工艺为传统的湿法制粒工艺,简单易行,通过多次实验研究可知发明的处方工艺重现性好,能充分保证产品批间质量的一致性。
附图说明
25.图1为实施例1和原研药在ph6.8磷酸盐缓冲液溶出介质的溶出曲线对比图;
26.图2为实施例1和原研药在ph6.0磷酸氢二钠-枸橼酸缓冲液溶出介质的溶出曲线对比图;
27.图3为实施例1和原研药在ph5.0醋酸盐缓冲液溶出介质的溶出曲线对比图;
28.图4为实施例1和原研药在水溶出介质的溶出曲线对比图;
29.图5为实施例1和原研药在ph1.2盐酸溶液溶出介质的溶出曲线对比图;
30.图6为实施例2~4和原研药在ph6.8磷酸盐缓冲液溶出介质的溶出曲线对比图;
31.图7为实施例2~4和原研药在ph6.0磷酸氢二钠-枸橼酸缓冲液溶出介质的溶出曲线对比图;
32.图8为实施例2~4和原研药在ph5.0醋酸盐缓冲液溶出介质的溶出曲线对比图;
33.图9为实施例2~4和原研药在水溶出介质的溶出曲线对比图。
具体实施方式
34.下面结合附图和具体实施方式对本发明作进一步详细说明。
35.本发明提供了一种克拉霉素片的制备方法,该克拉霉素的基本处方如表1所示;本发明所用的设备和仪器如表2所示。
36.表1本发明的基本处方
37.名称用量/1万片百分比克拉霉素2500.0g48.9%微晶纤维素1030.7g20.1%羟丙纤维素309.3g6.0%预胶化淀粉773.3g15.1%交联羧甲基纤维素钠257.3g5.0%司盘-806.7g0.1%聚维酮k30131.1g2.6%二氧化硅15.5g0.3%硬脂酸51.6g1.0%硬脂酸镁41.2g0.8%黄色包衣粉153.3g
‑‑‑
38.表2本发明实施例中所用的设备和仪器
[0039][0040][0041]
实施例1:
[0042]
本实施例为中试放大实验,通过本实施例考察处方工艺的可行性和重现性,本实施例所采用的处方如表3所示,试验批量为5.0万片/批。
[0043]
表3实施例1的处方
[0044][0045]
本实施例的制备方法包括如下步骤:
[0046]
s1、原辅料预处理:将克拉霉素原料、预胶化淀粉、微晶纤维素、交联羧甲基纤维素
钠和羟丙纤维素分别过60目筛;
[0047]
s2、配制增溶剂和粘合剂:
[0048]
向司盘80溶液中加入95%乙醇,搅拌均匀,配制成浓度为10%的司盘-80醇溶液;以1.0kg司盘-80醇溶液为例,称取0.1kg司-80溶液,加入0.9kg 95%乙醇,搅拌均匀即可;
[0049]
将聚维酮k30加入纯化水中,搅拌溶解,制得浓度为8%的聚维酮k30粘合剂;以10kg聚维酮k30粘合剂为例,称取0.8kg聚维酮k30,加入9.2kg纯化水,搅拌溶解即得;
[0050]
s3、制粒:将处方量的预胶化淀粉、微晶纤维素和交联羧甲基纤维素钠投入湿法制粒机中,开启搅拌桨,混合时间为2min、搅拌桨转速为180~270rpm,切割刀转速为1500-2000rpm;然后加入10%司盘-80醇溶液,每1万片料加65-70g司盘-80醇溶液,搅拌桨转速为50~100rpm,切割刀转速为1500-2000rpm;全部加入后搅拌桨运行3分钟,搅拌桨转速为180~270rpm,继续搅拌均匀;加入处方量的克拉霉素原料和羟丙纤维素,搅拌桨转速为180~270rpm、切割刀转速为1500-2000rpm的条件下混合3分钟,搅拌均匀后每1万片加入1600-1700g聚维酮k30粘合剂在同样的转速下继续搅拌;制粒6~15分钟后,在800-1500rpm转速下用375q不锈钢筛进行湿整粒后干燥,进风温度为70~80℃、进风风量为500~1500m3/h;当物料温度达到42~47℃、干燥失重在2.0~3.5%时,即可出料;
[0051]
s4、整粒、总混:用整粒机对干燥后的颗粒进行整粒,筛网型号为062g,整粒刀为方刀,整粒速率为800~1500rpm,整粒过程中加入处方量的二氧化硅、硬脂酸和硬脂酸镁,整粒后的颗粒转移到混合机中进行总混,混合时间为5-10min,转速为10rpm,得到总混颗粒,总混颗粒的干燥失重为1.5~4.0%;
[0052]
s5、压片:将总混颗粒投料至压片机进行压片,压片参数如下:模具规格为15.5*7.8mm浅凹椭圆形冲、硬度为9.0~15.0kg、片厚为5.3~5.9mm、片重差异为预压片重的
±
4.5%、脆碎度:不得有裂片或缺边,得到素片;对素片进行薄膜包衣,得到克拉霉素片;
[0053]
s6、包衣、铝塑包装
[0054]
对素片进行薄膜包衣,包衣参数如下:包衣液为浓度为8%的黄色包衣液(使用75%乙醇配制),片床温度为37~45℃,包衣增重理论上为3.0%。
[0055]
将包衣片进行铝塑包装,内包装材料为pvc和铝箔。
[0056]
为了对比本实施例与原研药(商品名为klacid,规格为250mg,生产商为意大利abbvie s.r.l.,在意大利上市,产品批号1057203)的溶出曲线,将实施例1所制得的克拉霉素片和原研药在不同的溶出介质中测定溶出曲线,具体结果如下:
[0057]
一、溶出介质为ph6.8磷酸盐缓冲液的溶出曲线
[0058]
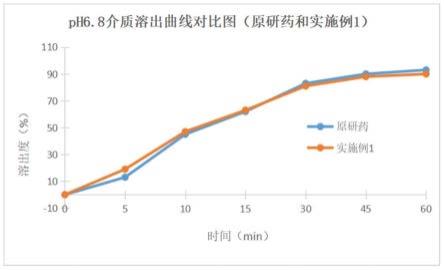
以ph6.8磷酸盐缓冲液作为溶出介质测定溶出曲线,克拉霉素片在该介质下溶解度为约0.5mg/ml,克拉霉素片在该介质中呈缓慢溶出,是较有区分力的一条溶出曲线,对比原研药及实施例1的溶出曲线,结果如图1和表4所示:
[0059]
表4实施例1与原研药在ph6.8磷酸盐缓冲液中的溶出曲线
[0060]
[0061]
由图1和表4可知,实施例1所制得的克拉霉素片与原研药相比,溶出曲线相似度较高,f2相似因子为84,说明实施例1所制得的克拉霉素片与原研药在磷酸盐缓冲液(ph6.8)中的溶出曲线具有较好相似性。
[0062]
二、溶出介质为磷酸氢二钠-枸橼酸缓冲液(ph6.0)的溶出曲线
[0063]
磷酸氢二钠-枸橼酸缓冲液(ph6.0)是日本橙皮书中使用的溶出介质。以磷酸氢二钠-枸橼酸缓冲液(ph6.0)作为溶出介质测定溶出曲线,克拉霉素片在该介质下溶解度约为1.5mg/ml,对比原研药及实施例1的溶出曲线,结果如图2和表5所示:
[0064]
表5实施例1与原研药在磷酸氢二钠-枸橼酸缓冲液(ph6.0)中的溶出曲线
[0065][0066][0067]
由图2和表5可知,在磷酸氢二钠-枸橼酸缓冲液(ph6.0)的溶出介质中,实施例1所制得的克拉霉素片与原研药的溶出度在15分钟均达到85%以上,不需比较f2值,可认为该溶出曲线区分力不强,实施例1所制得的克拉霉素片与原研药在该介质下的溶出曲线相似。
[0068]
三、溶出介质为醋酸盐缓冲液(ph5.0)的溶出曲线
[0069]
醋酸盐缓冲液(ph5.0)是中国药典及美国fda溶出数据库中使用的溶出介质,以其作为溶出介质测定溶出曲线,克拉霉素在该介质下溶解度约为9mg/ml,溶解度较大,对比原研药及实施例1的溶出曲线,结果如图3和表6所示:
[0070]
表6实施例1与原研药在醋酸盐缓冲液(ph5.0)中的溶出曲线
[0071][0072]
由图3和表6可知,在醋酸盐缓冲液(ph5.0)的溶出介质中,实施例1所制得的克拉霉素片与原研药的溶出度均在15分钟达到85%以上(10分钟已基本达到顶峰),不需比较f2值,可认为该溶出曲线区分力不强,实施例1所制得的克拉霉素片与原研药在该介质下的溶出曲线相似。
[0073]
四、溶出介质为水的溶出曲线
[0074]
水中的溶出曲线也是日本橙皮书中4条溶出曲线之一,克拉霉素片在水中几乎不溶,测定的溶解度仅为约0.02mg/ml,克拉霉素片250mg在900ml水中仅能溶解10%左右,对比原研药及实施例1在水中的溶出曲线,结果如图4和表7所示:
[0075]
表7实施例1与原研药在水中的溶出曲线
[0076][0077]
由图4和表7可知,在水作为溶出介质的条件下,实施例1所制得的克拉霉素片与原研药均不能很好溶出,在15分钟后基本达到平台期,溶出度约为10%左右,溶液已达饱和状态,可见实施例1所制得的克拉霉素片与原研药在水中的溶出状态一致。
[0078]
五、溶出介质为盐酸溶液(ph1.2)的溶出曲线
[0079]
克拉霉素在ph1.2的介质中会分解,因而其溶出过程存在一个边释放边降解的动态过程。对比原研药及实施例1在盐酸溶液(ph1.2)中的溶出曲线,结果如图5和表8所示:
[0080]
表8实施例1与原研药在盐酸溶液(ph1.2)中的溶出曲线
[0081][0082]
由图5和表8可知,克拉霉素片在ph1.2作为溶出介质的条件下,并不像在其他介质中那样存在崩解释放的过程,而是慢慢的溶释状态,实施例1所制得的克拉霉素片与原研药在盐酸溶液(ph1.2)中的溶出现象上较相似。在该溶出条件下,慢慢释放出来的克拉霉素会与ph1.2盐酸溶液反应而分解,对于溶出度的测定影响较大,因此不建议选择该介质作为溶出介质比较一致性。
[0083]
由实施例1所制得的克拉霉素片在不同介质中的溶出曲线测定结果可知,按照本发明所制得的克拉霉素片与原研药在ph6.8、ph6.0、ph5.0、ph1.2和水五种介质中溶出曲线相似且在区分力最大的介质(ph6.8)中相似因子能够大于72,相似度较高,说明本发明所制得的克拉霉素片和原研药的体外溶出一致性较好。
[0084]
实施例2~4:
[0085]
实施例2~4为进一步的中试放大实验,通过实施例2~4考察处方工艺的可行性和重现性,实施例2~4所采用的处方如表9所示,每个实施例的试验批量为15.0万片/批。
[0086]
表9实施例2的处方
[0087][0088]
实施例2~4的制备方法与实施例1相同。
[0089]
对实施例2~4所制得的克拉霉素片与原研药进行成品检测,结果如表10所示:
[0090]
表10实施例2~4与原研药的成品检测结果
[0091]
[0092][0093]
为了对比实施例2~4与原研药(商品名为klacid,规格为250mg,生产商为意大利abbvie s.r.l.,在意大利上市,产品批号1057203)的溶出曲线,将实施例2~4所制得的克拉霉素片和原研药在不同的溶出介质中测定溶出曲线,具体结果如下:
[0094]
一、溶出介质为ph6.8磷酸盐缓冲液的溶出曲线
[0095]
表11实施例2~4与原研药在ph6.8磷酸盐缓冲液中的溶出曲线
[0096][0097]
表11为实施例2~4与原研药在磷酸盐缓冲液(ph6.8)中的溶出数据结果,由图6和表11可知,实施例2~4与原研药相比较,在ph6.8磷酸盐缓冲液介质中,溶出曲线相似度分别为78、58和63,相似度均较高,说明实施例2~4与原研药在在磷酸盐缓冲液(ph6.8)中的溶出曲线具有高度相似性。
[0098]
二、溶出介质为磷酸氢二钠-枸橼酸缓冲液(ph6.0)的溶出曲线
[0099]
表12实施例2~4与原研药在磷酸氢二钠-枸橼酸缓冲液(ph6.0)中的溶出曲线
[0100][0101]
表12为实施例2~4与原研药在磷酸盐缓冲液(ph6.8)中的溶出数据结果,由图7和表12可知,在磷酸氢二钠-枸橼酸缓冲液(ph6.0)溶出介质中,实施例2~4与原研药的溶出度均在15分钟达到85%以上,不需比较f2值,可认为该溶出曲线区分力不强,实施例2~4与原研药在该介质下溶出曲线相似。
[0102]
三、溶出介质为醋酸盐缓冲液(ph5.0)的溶出曲线
[0103]
表13实施例2~4与原研药在醋酸盐缓冲液(ph5.0)中的溶出曲线
[0104][0105]
由图8和表13可知,在醋酸盐缓冲液(ph5.0)的溶出介质中,实施例2~4所制得的克拉霉素片与原研药的溶出度均在15分钟达到85%以上(10分钟已基本达到顶峰),不需比较f2值,可认为该溶出曲线区分力不强,实施例2~4所制得的克拉霉素片与原研药在该介质下的溶出曲线相似。
[0106]
四、溶出介质为水的溶出曲线
[0107]
表14实施例2~4与原研药在水中的溶出曲线
[0108][0109]
由图9和表13可知,在水作为溶出介质的条件下,实施例2~4所制得的克拉霉素片与原研药均不能很好溶出,在15分钟后基本达到平台期,溶出度约为10%左右,溶液已达饱
和状态,可见实施例2~4所制得的克拉霉素片与原研药在水中的溶出状态一致。
[0110]
五、样品加速稳定性考察
[0111]
1、溶出曲线
[0112]
以实施例2作为样品,测定不同加速条件下的溶出曲线。由于ph6.8磷酸盐缓冲液介质为最有区分力的介质,加速样品检测实验优先检测该介质的溶出曲线,结果如表15所示:
[0113]
表15实施例2在不同加速条件下的溶出曲线
[0114][0115]
由表15可知,实施例2在加速考察后ph6.8磷酸盐缓冲液介质中的溶出曲线有变快的趋势,但变快幅度较不明显,溶出曲线变快的主要原因为样品加速过程中吸水引起包衣片硬度下降,最终导致溶出曲线前期溶出变快,但从总的趋势来看,样品加速前后溶出曲线稳定性较好。
[0116]
2、有关物质检测
[0117]
表16实施例2在不同加速条件下的有关物质检测结果
[0118][0119]
由表16可知,实施例2在加速考察后样品有关物质项目与0天相比均无明显增长,样品加速前后有关物质稳定性好。
[0120]
3、含量检测
[0121]
表17实施例2在不同加速条件下的含量检测结果
[0122]
[0123]
由表17可知,实施例2在加速考察后样品含量项目与0天相比均无明显变化趋势,样品加速前后有关物质稳定性好。
[0124]
由实施例2~4所制得的克拉霉素片在不同介质中的溶出曲线测定结果可知,按照本发明所制得的克拉霉素片与原研药在ph6.8、ph6.0、ph5.0和水四种介质中溶出曲线相似且在区分力最大的介质(ph6.8)中相似因子能够大于72,相似度较高,说明本发明所制得的克拉霉素片和原研药的体外溶出一致性较好。且从实施例2加速稳定性考察后的关键质量检测数据来看,区分力最大介质中的溶出曲线、有关物质和含量的稳定性均较好,说明本发明所制得产品的质量稳定。
[0125]
对比例1:
[0126]
本对比例与实施例1的区别仅在于粘合剂的配制方式不同,具体如下:
[0127]
s2、配制粘合剂:
[0128]
称取0.004kg司盘80溶液中加入0.482kg95%乙醇,搅拌均匀后再加入0.434kg纯化水,最后再称取0.08kg聚维酮k30加入其中,搅拌溶解即可;
[0129]
s3、制粒:将处方量的克拉霉素原料、羟丙纤维素、预胶化淀粉、微晶纤维素和交联羧甲基纤维素钠投入湿法制粒机中,在搅拌桨转速为180~270rpm、切割刀转速为1500-2000rpm的条件下,混合时间2分钟。加入步骤s2所制得的粘合剂,每1万片加粘合剂1600-1700g,搅拌桨转速为180~270rpm,切割刀转速为1500-2000rpm,制粒时间为6~15分钟。
[0130]
在上述制粒过程中,湿颗粒中结团较多,容易导致所制颗粒粗细不均匀,影响颗粒的流动性,原因在于原料或某种辅料与司盘80接触后容易团聚结块。
[0131]
其他操作均与实施例1相同。
[0132]
为了对比本对比例与原研药(商品名为klacid,规格为250mg,生产商为意大利abbvie s.r.l.,在意大利上市,产品批号1057203)的溶出曲线,将对比例1所制得的克拉霉素片和原研药在ph6.8磷酸盐缓冲液的溶出介质中测定溶出曲线,具体结果如表18所示:
[0133]
表18对比例1与原研药在ph6.8磷酸盐缓冲液中的溶出曲线
[0134][0135]
由表18可知,司盘80溶于聚维酮k30溶液中的方式没能达到增溶效果,反而引起了湿颗粒结团和制粒不均匀,导致溶出曲线明显差于原研药。
[0136]
对比例2:
[0137]
本对比例与实施例1的区别在于司盘-80醇溶液的加入方式不同,具体如下:
[0138]
s2、配制粘合剂:
[0139]
称取0.08kg聚维酮k30,加入0.92kg纯化水,搅拌溶解即得;
[0140]
s3、制粒:将处方量的克拉霉素原料、羟丙纤维素、预胶化淀粉、微晶纤维素和交联羧甲基纤维素钠投入湿法制粒机中,在搅拌桨转速为180~270rpm、切割刀转速为1500-2000rpm的条件下,混合时间2分钟。加入步骤s2所制得的8%聚维酮k30粘合剂:每1万片加聚维酮k30粘合剂1600-1700g,搅拌桨转速为180~270rpm,切割刀转速为1500-2000rpm,制
粒时间为6~15分钟。
[0141]
粘合剂中不添加司盘80制粒后湿颗粒较均匀,去除司盘80后对制粒工艺无明显影响。
[0142]
s6、包衣
[0143]
包衣液的配制:将80g黄色包衣粉加入到920g 75%乙醇中,搅拌过程中加入3.5g司盘80一起搅拌,搅拌至包衣粉全部分散后过筛备用。
[0144]
对素片进行薄膜包衣,包衣参数如下:包衣液为浓度为8%的黄色包衣液(使用75%乙醇配制),片床温度为37~45℃,包衣增重理论上为3.0%。
[0145]
其他操作均与实施例1相同。
[0146]
为了对比本对比例与原研药(商品名为klacid,规格为250mg,生产商为意大利abbvie s.r.l.,在意大利上市,产品批号1057203)的溶出曲线,将对比例2所制得的克拉霉素片和原研药在ph6.8磷酸盐缓冲液的溶出介质中测定溶出曲线,具体结果如表19所示:
[0147]
表19对比例2与原研药在ph6.8磷酸盐缓冲液中的溶出曲线
[0148][0149]
由表19可知,司盘80加入到包衣粉中后,包衣片的包衣膜在ph6.8磷酸盐缓冲液中破膜时间缩短,导致5分钟时间点溶出度提高,5分钟点溶出度明显快于原研药,原因在于将司盘80加入到包衣液中会导致包衣膜在缓冲液中溶解的速度加快,从而导致包衣膜破膜时间缩短。片芯不加司盘80后,10分钟后续时间点溶出度相比原研药明显偏慢,原因在于少了司盘80的增溶作用导致原料溶出速度变慢。
[0150]
上述实施方式仅为本发明的优选实施方式,不能以此来限定本发明保护的范围,本领域的技术人员在本发明的基础上所做的任何非实质性的变化及替换均属于本发明所要求保护的范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。