1.本发明涉及即便在吸收液体后加压下的液体回流也少的颗粒状吸水剂。
背景技术:
2.吸水性树脂(sap/superabsorbentpolymer)是水溶胀性且水不溶性的高分子胶凝剂。以吸水性树脂作为主成分的颗粒状吸水剂被利用于纸尿布、生理用卫生巾、面向成人的失禁用制品等卫生物品、农林园艺用的土壤保水剂、工业用的止水剂等各种用途的吸收物品中。作为这种吸水性树脂的原料,提出了多种单体、亲水性高分子,但从性能和成本的观点出发,将丙烯酸和/或其盐用作单体的聚丙烯酸(盐)系吸水性树脂最为常用。
3.随着颗粒状吸水剂的主要用途即纸尿布的高性能化,对颗粒状吸水剂要求多种功能(物性)。作为颗粒状吸水剂的物性的具体例,不限定于单纯的吸水倍率高,还可列举出凝胶强度、水可溶成分、吸水速度、加压下的吸水倍率、通液性、粒度分布、耐尿性、抗菌性、耐冲击性(耐损伤性)、粉体流动性、消臭性、耐着色性(白色度)、低粉尘等。这些之中,有从颗粒状吸水剂中朝着被吸收的液体的导入方向再次释放出液体的所谓“倒流”物性。倒流会因接触肌肤的液体而导致使用者感到不适,因此,降低倒流在吸水性树脂的领域中是非常重要的课题(例如专利文献1)。
4.需要说明的是,作为相关的其它现有技术,有专利文献2。
5.现有技术文献
6.专利文献
7.专利文献1:日本特开2015-213911号公报
8.专利文献2:国际公开第97/003114号
技术实现要素:
9.然而,并不是每次对纸尿布等吸收物品排出尿等液体后就更换吸收物品,通常对吸收物品进行多次排液。吸收了液体的颗粒状吸水剂会呈现溶胀状态。因而,使用者有时会使用具有溶胀状态的颗粒状吸水剂的吸收物品。像这样,在颗粒状吸水剂为溶胀状态时,需要即便因日常动作、尤其是承载有体重的状态(例如仰卧状态、坐下等动作)等而从外部施加有压力的情况下,吸收体的液体保持力也会提高,液体回流也会降低,由此,也可降低皮肤发炎等烦扰,使用者能够长期舒适地使用吸收物品。以往仅关注到通常用加压下的吸收倍率(aap)来表现的“在承载有载荷的条件下如何吸收液体”,并未着眼于在吸液后的溶胀状态下即便承载有载荷也能够保持多少液体,可以认为尚有改良的余地。
10.因而,本发明的目的在于,提供:在颗粒状吸水剂为溶胀状态时,即便颗粒状吸水剂从外部负载有压力,也能够显著降低液体回流的颗粒状吸水剂。
11.通过如下的颗粒状吸水剂来解决上述课题,所述颗粒状吸水剂以经表面交联而成的聚丙烯酸(盐)系吸水性树脂作为主成分,所述颗粒状吸水剂满足下述式(1)。
12.aap(2.06kpa) rcap(2.06kpa)≥0.58
×
crc 55.6(1)
13.(式(1)中,aap(2.06kpa)表示在加压2.06kpa条件下的吸水倍率(g/g),rcap(2.06kpa)表示在溶胀后且加压下的吸水倍率(g/g),crc表示未加压下的吸水倍率(g/g))。
附图说明
14.图1是表示用于测定凝胶渗透速度(gpr)的装置的示意图。
15.图2是表示在各实施例和比较例中,对crc[g/g](横轴)标绘aap(2.06kpa) rcap(2.06kpa)[g/g](纵轴)而得到的图。直线表示aap(2.06kpa) rcap(2.06kpa)=0.58
×
crc 55.6。
具体实施方式
[0016]
以下,边示出最佳方式边说明本发明。对于本说明书的整体而言,只要没有特别提及,则单数形式的表达应该理解为还包括其复数形式的概念。因此,只要没有特别提及,则应该理解为单数形式的冠词(例如英语情况下的“a”、“an”、“the”等)还包括其复数形式的概念。另外,只要没有特别提及,则本说明书中使用的术语应该理解为按照该领域中通常使用的含义来使用。因此,只要没有另行定义,则本说明书中使用的全部专业术语和科学技术术语具有与本发明所属领域的技术人员通常理解的含义相同的含义。发生矛盾的情况下,本说明书(包括定义在内)优先。本发明不限定于下述实施方式,可以在权利要求书的范围内进行各种变更。
[0017]
〔1〕术语的定义
[0018]
(1-1)“吸水性树脂”[0019]
本发明中的“吸水性树脂”是指水溶胀性且水不溶性的高分子交联体,其满足以下的物性。即,是指如下高分子交联体:作为“水溶胀性”,满足ert441.2-02中规定的crc为5g/g以上的物性,并且,作为“水不溶性”,满足ert470.2-02中规定的ext为50重量%以下的物性。
[0020]
上述吸水性树脂可根据其用途来适当设计,没有特别限定,优选为使具有羧基的不饱和单体发生交联聚合而得到的亲水性交联聚合物。另外,不限定于总量(100重量%)为聚合物的形态,可以是在满足上述物性(crc、ext)的范围内包含添加剂等的吸水性树脂组合物。
[0021]
进而,本发明中的吸水性树脂不限定于最终制品,有时也指代吸水性树脂的制造工序中的中间体(例如聚合后的含水凝胶状交联聚合物、干燥后的干燥聚合物、表面交联前的吸水性树脂粉末等),包括它们中的全部在内,统称为“吸水性树脂”。需要说明的是,作为吸水性树脂的形状,可列举出片状、纤维状、薄膜状、颗粒状、凝胶状等,本发明的吸水性树脂以颗粒状(粉末)为主。
[0022]
(1-2)“颗粒状吸水剂”[0023]
本说明书中,吸水剂包含吸水性树脂作为主成分。本说明书中,颗粒状吸水剂是指颗粒状(别称:粉末状)的吸水剂(包含吸水性树脂颗粒作为主成分),无论是单粒的颗粒状吸水剂还是多个的颗粒状吸水剂均称为颗粒状吸水剂。“颗粒状”是指具有颗粒的形态,颗粒是指具有能够测定的大小且呈现固态或液态的粒状小物体(jis工业术语大辞典第4版、2002页)。需要说明的是,本说明书中,有时也将颗粒状吸水剂简称为吸水剂。
[0024]
需要说明的是,水性液体不限定于水,可以是尿、血液、汗、粪、废液、湿气、蒸气、冰、水与有机溶剂和/或无机溶剂的混合物、雨水、地下水等,只要包含水就没有特别限定。可优选列举出尿、经血、汗、其它体液。
[0025]
本发明所述的颗粒状吸水剂可适合地用作用于吸收水性液体的卫生材料。本发明的颗粒状吸水剂以经表面交联而成的聚丙烯酸(盐)系吸水性树脂(颗粒)(以下也简称为聚丙烯酸(盐)系吸水性树脂)作为主成分。换言之,在颗粒状吸水剂中,经表面交联而成的聚丙烯酸(盐)系吸水性树脂的含量优选为60~100重量%、70~100重量%、80~100重量%、90~100重量%。另外,颗粒状吸水剂任选包含其它吸水性树脂颗粒、水、和/或、水不溶性无机颗粒、水溶性的含有多价金属阳离子的化合物等添加剂。颗粒状吸水剂的适合含水率为0.2~30重量%。即,这些成分经一体化而成的吸水性树脂组合物也属于颗粒状吸水剂的范畴。
[0026]
需要说明的是,吸水剂中的聚丙烯酸(盐)系吸水性树脂的上限为99重量%,进而为97重量%,特别为95重量%左右,优选还包含水、后述的添加剂(水不溶性无机颗粒、水溶性的含有多价金属阳离子的化合物)。
[0027]
另外,本发明的颗粒状吸水剂中,以聚丙烯酸(盐)系吸水性树脂作为主成分,颗粒状吸水剂可以含有其它的吸水性树脂。作为其它的吸水性树脂,可列举出聚磺酸(盐)系吸水性树脂、马来酸酐(盐)系吸水性树脂、聚丙烯酰胺系吸水性树脂、聚乙烯醇系吸水性树脂、聚环氧乙烷系吸水性树脂、聚天冬氨酸(盐)系吸水性树脂、聚谷氨酸(盐)系吸水性树脂、聚藻酸(盐)系吸水性树脂、淀粉系吸水性树脂、纤维素系树脂等。
[0028]
(1-3)“聚丙烯酸(盐)”和“聚丙烯酸(盐)系吸水性树脂”[0029]
本发明中的“聚丙烯酸(盐)”是指聚丙烯酸和/或其盐。另外,聚丙烯酸(盐)系吸水性树脂是指:作为主成分,包含丙烯酸和/或其盐(以下称为“丙烯酸(盐)”)作为重复单元,优选利用接枝成分进行了内部交联的聚丙烯酸(盐)经表面交联而成的树脂。
[0030]
聚丙烯酸(盐)系吸水性树脂在颗粒状吸水剂中优选为颗粒状(别称:粉末状)。
[0031]
需要说明的是,上述“主成分”是指:丙烯酸(盐)的用量(含量)相对于聚合中使用的单体(不包括内部交联剂在内)整体,通常为50~100摩尔%、优选为70~100摩尔%、更优选为90~100摩尔%、进一步优选实质为100摩尔%。
[0032]
(1-4)“edana”和“ert”[0033]“edana”是欧州非织造布工业协会(european disposables and nonwovens associations)的简称,“ert”是欧州标准(基本为世界标准)的吸水性树脂的测定方法(edana recommended test methods)的简称。本发明中,只要没有特别记载,则按照ert原本(2002年修订/公知文献)来测定吸水性树脂的物性。
[0034]
(1-5)“psd”(ert420.2-02)
[0035]“psd”是粒度分布(particle size distribution)的简称,是指通过筛分级而测得的颗粒状吸水剂或吸水性树脂的粒度分布。
[0036]
需要说明的是,重均粒径(d50)和粒度分布的对数标准偏差(σζ)利用与美国专利第7638570号中记载的“(3)mass-average particle diameter(d50)and logarithmic standard deviation(σζ)of particle diameter distribution”相同的方法进行测定。
[0037]
(1-6)其它
[0038]
本说明书中,表示范围的“x~y”是指“x以上且y以下”。另外,只要没有特别注释,则重量的单位“t(吨)”是指“公吨(metric ton)”,“ppm”是指“重量ppm”或“质量ppm”。进而,“重量”与“质量”、“重量份”与“质量份”、“重量%”与“质量%”分别视作同义词。另外,“~酸(盐)”是指“~酸和/或其盐”,“(甲基)丙烯酰基”是指“丙烯酰基和/或甲基丙烯酰基”。
[0039]
另外,有时将“升”简写为“l”或“l”,将“重量%”简写为“wt%”。进而,在进行微量成分的测定时,将检出限以下表述为n.d(non detected)。
[0040]
〔2〕颗粒状吸水剂
[0041]
本发明的颗粒状吸水剂以经表面交联而成的聚丙烯酸(盐)系吸水性树脂作为主成分,所述颗粒状吸水剂满足式(1)。
[0042]
本发明的颗粒状吸水剂在颗粒状吸水剂为溶胀状态时,即便从外部对颗粒状吸水剂施加有压力,也能够显著降低液体回流。
[0043]
以下,也将利用aap(2.06kpa) rcap(2.06kpa)而求出的值称为值(a),将利用0.58
×
crc 55.6而求出的值称为值(b)。
[0044]
颗粒状吸水剂不满足式(1)、即值(a)《值(b)时,颗粒状吸水剂吸水而溶胀后,液体回流量显著变大。
[0045]
值(a)是aap(2.06kpa)与rcap(2.06kpa)之和。“aap”是absorption against pressure的简称,是指颗粒状吸水剂在加压下的吸水倍率。“rcap”是retention capacity against pressure的简称,是指颗粒状吸水剂发生溶胀时的在加压下的吸水倍率。另外,“crc”是centrifuge retention capacity(离心分离机保持容量)的简称,是指颗粒状吸水剂或吸水性树脂在未加压下的吸水倍率(有时也称为“吸水倍率”)。通常,吸水倍率在加压下发生降低,因此,在同一颗粒状吸水剂中,aap(2.06kpa)[g/g]《crc(2.06kpa)[g/g]。
[0046]
在设想吸水性物品的实际使用时,可联想到各种使用状况,例如在承载有体重的状态下进行排尿的情况、排尿时在无加压条件下发生溶胀且随着动作而被加压的情况等各种场合。本发明人等在想要解决“颗粒状吸水剂发生吸水而溶胀后的回流量降低”这一课题的过程中发现:各种场合中的“液体保持力”(吸水性树脂颗粒自身的吸水倍率(即crc)加上在吸水性树脂颗粒彼此(包括一次颗粒和/或一次颗粒彼此聚集而成的二次颗粒)的间隙中保持的液体量而得到的吸收量)是重要的,在颗粒状吸水剂的多种物性中,着眼于aap(2.06kpa)和rcap(2.06kpa)。
[0047]
此处,对于“颗粒状吸水剂为溶胀状态时,即便从外部对颗粒状吸水剂施加压力也可显著降低液体回流”这一本技术课题而言,可以认为如果rcap高则可解决课题。然而,在本发明人等的研究过程中已明确:对于溶胀状态的液体回流而言,仅考虑溶胀时的加压下的吸水倍率即rcap并不充分。例如,根据后述比较例1-5的rcap高于实施例1-8,但溶胀时的颗粒状吸水剂的液体回流显著降低也可理解该事实。并且,本发明人等发现:aap(2.06kpa) rcap(2.06kpa)的总和以及crc是与溶胀时的颗粒状吸水剂的液体回流有关的重要因素,进而发现:满足式(1)的新型颗粒状吸水剂可显著抑制溶胀时的颗粒状吸水剂的液体回流。
[0048]
需要说明的是,通常作为吸水性树脂的物性评价而使用的“倒流”(有时也称为回流量、re-wet)是针对将包含吸水性树脂(吸水剂)、纸浆等的吸收层用非织造布等层叠而得到的吸收片(吸收体)进行的评价,并不是吸水性树脂(吸水剂)自身的评价。另外,在前述re-wet评价中,无法说吸收体中包含的吸水性树脂呈现饱和状态(在吸收体表面上以30分
钟的间隔多次吸收液体后,在载荷下评价回流),并没有评价本技术课题、即“溶胀状态”的颗粒状吸水剂在进一步加压的状态下的液体保持力。
[0049]
另外,颗粒状吸水剂更优选满足以下式(2)。
[0050]
aap(2.06kpa) rcap(2.06kpa)≥0.58
×
crc 56.0(2)
[0051]
通过为满足上述式(2)的颗粒状吸水剂,从而对已溶胀的吸收体进行加压时的液体保持力优异。
[0052]
尤其是,颗粒状吸水剂的crc小于37.0g/g时,更优选满足上述式(2)。
[0053]
进而,颗粒状吸水剂更优选满足以下式(3)。
[0054]
aap(2.06kpa) rcap(2.06kpa)≥0.58
×
crc 56.5(3)
[0055]
通过为满足上述式(3)的颗粒状吸水剂,从而对已溶胀的吸收体进行加压时的液体保持力优异。
[0056]
尤其是,颗粒状吸水剂的crc小于37.0g/g时,更优选满足上述式(3)。
[0057]
作为值(a),没有特别限定,优选大于76.0g/g。即,颗粒状吸水剂优选满足下述式(a)。
[0058]
aap(2.06kpa) rcap(2.06kpa)》76.0(a)
[0059]
通过使值(a)大于76.0g/g,从而容易进一步发挥出颗粒状吸水剂的溶胀后的液体回流的降低。值(a)的上限值没有特别限定,通常为90.0g/g以下,可以为85.0g/g以下。
[0060]
需要说明的是,在计算式(1)~(3)和式(a)时,aap(2.06kpa)、rcap(2.06kpa)和crc使用截止到小数点后第一位的测定值,在计算值(a)、值(b)、以及式(2)和式(3)的右边时,将使用上述测定值而算出的值的小数点后第二位四舍五入,使用截止到小数点后第一位的值。
[0061]
(2-1)crc(离心分离机保持容量)(ert441.2-02)
[0062]“crc”是centrifugeretentioncapacity(离心分离机保持容量)的简称,是指颗粒状吸水剂或吸水性树脂的未加压下的吸水倍率(有时也称为“吸水倍率”)。
[0063]
具体而言,是指:将颗粒状吸水剂或吸水性树脂0.2g装入至非织造布制的袋后,在明显过量的0.9重量%氯化钠水溶液中浸渍30分钟而使其自由溶胀,其后,利用离心分离机(250g)进行控水后的吸水倍率(单位:g/g)。
[0064]
本发明的颗粒状吸水剂的crc(离心分离机保持容量)优选为30g/g以上、更优选为31g/g以上、进一步优选为32g/g以上、更进一步优选为33g/g以上。通过使crc为30g/g以上,从而吸收量变得适当,可确保作为纸尿布等卫生物品的吸收体而言的性能。另外,颗粒状吸水剂的crc(离心分离机保持容量)优选为70g/g以下、更优选为60g/g以下、进一步优选为50g/g以下、特别优选为40g/g以下。通过使crc为70g/g以下,从而保持吸收尿、血液等体液等的速度,因此,也适用于高吸水速度类型的纸尿布等。需要说明的是,crc可根据内部交联剂的种类、量等来控制。
[0065]
(2-2)加压下的吸水倍率(aap)(ert442.2-02)
[0066]“aap”是absorptionagainstpressure的简称,是指颗粒状吸水剂或吸水性树脂在加压下的吸水倍率。
[0067]
具体而言,aap(2.06kpa)是指:使颗粒状吸水剂或吸水性树脂0.9g相对于明显过量的0.9重量%氯化钠水溶液、在2.06kpa(21g/cm2、0.3psi)的载荷下溶胀1小时后的吸水
倍率(单位:g/g)。需要说明的是,有时也将载荷条件变更为4.83kpa(49g/cm2、0.7psi)来进行测定。该情况下,记作aap(4.83kpa)。
[0068]
另外,在ert442.2-02中也表述为absorption under pressure,但实质上内容相同。
[0069]
本发明的颗粒状吸水剂的aap(2.06kpa)优选为20g/g以上、更优选为24g/g以上、进一步优选为26g/g以上、更进一步优选为28g/g以上、特别优选为29g/g以上、最优选为30g/g以上。关于上限值,没有特别限定,优选为40g/g以下。通过满足上述条件,从而aap(2.06kpa)高至某种程度,因而容易满足式(1)的条件。另外,使用该颗粒状吸水剂而制造的纸尿布从纸浆中吸取尿的能力优异,能够降低回流量,能够抑制皮肤发炎、漏尿。需要说明的是,若参照后述实施例、比较例则可理解:aap(2.06kpa)高与满足式(1)没有相关性。
[0070]
另外,本发明的颗粒状吸水剂的aap(4.83kpa)优选为10g/g以上、更优选为13g/g以上、进一步优选为17g/g以上、特别优选为20g/g以上。关于上限值,没有特别限定,优选为30g/g以下。通过满足上述条件,从而使用上述颗粒状吸水剂而制造的纸尿布从纸浆中吸取尿的能力优异,能够降低回流量,能够抑制皮肤发炎、漏尿。需要说明的是,aap可利用表面交联剂的种类、量等来控制。
[0071]
(2-3)加压下的保持容量(retention capacity against pressure:rcap)
[0072]“rcap”是retention capacity against pressure的简称,是指颗粒状吸水剂在溶胀时且在加压下的吸水倍率。
[0073]
rcap的试验可以使用美国专利第8269060号说明书中记载的gel bed permeability(凝胶床透过性)试验所使用的料筒、活塞和重物来测定。具体而言,使用美国专利第8269060号说明书的图1中示出的装置。
[0074]
在实施试验之前,测定料筒、活塞和重物的总重量,将其值设为wa[g]。量取试验样品约0.9g,均匀地散布在料筒底面,在试验溶液(0.9重量%的氯化钠水溶液)中浸渍60分钟,使样品发生溶胀而不施加限制载荷。在60分钟后,对料筒中的样品承载活塞和重物(总重量约596g),将料筒从试验溶液中向上提起,放置在网眼为2000μm的jis标准筛上,进行约1分钟的控液。在控液后,用kimwipes(nippon paper crecia公司制的s-200)等将附着于料筒下部的水滴去除,进行重量测定,将所得值设为wb[g]。需要说明的是,利用kimwipes而进行的附着于料筒下部的水滴的去除如下进行:以不去除料筒中的溶胀凝胶层所包含的水分的方式,只不过是为了去除附着于料筒外的水分,以不受到kimwipes施加的压力的方式,在0.5秒以内进行擦拭。利用以下的式(4)来计算rcap。
[0075]
[数1]
[0076][0077]
本发明的颗粒状吸水剂的rcap(2.06kpa)优选为18g/g以上、更优选为24g/g以上、更进一步优选为30g/g以上、特别优选为40g/g以上、最优选为43g/g以上。关于上限值,没有特别限定,优选为60g/g以下。通过满足上述条件,从而rcap(2.06kpa)高至某种程度,因而容易满足式(1)的条件。另外,使用该颗粒状吸水剂而制造的纸尿布从纸浆中吸取尿的能力优异,能够降低回流量,能够抑制皮肤发炎、漏尿。需要说明的是,如果参照后述实施例、比较例则可理解:rcap(2.06kpa)高与满足式(1)没有相关性。
[0078]
rcap(2.06kpa)可通过控制吸水性树脂的制造方法、例如添加剂(例如水不溶性无机颗粒)的混合时间、可溶成分的含量来控制其数值。
[0079]
(2-3)凝胶渗透速度(gel permeation rate:gpr)
[0080]
本说明书中,颗粒状吸水剂的“通液性”是指在载荷下在溶胀凝胶颗粒之间通过的液体的流动性。作为该指标,可使用凝胶渗透速度(gpr)。颗粒状吸水剂的gpr测定通过参考美国专利第5849405号说明书中记载的食盐水流动诱导性(sfc)试验,变更测定条件并按照以下的步骤来进行。
[0081]
作为用于测定的装置,使用图1中示出的装置400。装置400大体上由容器410和罐420构成。在容器410中设置有盒(cell)411(内径为6cm),在盒411的内部能够收纳溶胀凝胶414(使颗粒状吸水剂吸水而得到的物质),并且能够导入液体423。另外,通过使活塞412嵌合于盒411,从而能够对溶胀凝胶414施加压力。盒411的底面和活塞412的底面铺设有金属网413a、413b(no.400不锈钢制的金属网、网眼为38μm),使得溶胀凝胶414(和颗粒状吸水剂)无法通过。此处,液体423使用0.90质量%的氯化钠水溶液。罐420在内部蓄积有液体423。液体423通过带有旋塞的l字管422而导入至盒411中。另外,罐420中插入有玻璃管421,玻璃管421的内部被空气充满。由此,能够使玻璃管421的下端与盒411内的液面相同。即,在罐420内的液体423的液面位于玻璃管421下端的上方的期间内,能够将盒411内的液面保持恒定。在本次的测定中,将罐420内的液体423的下部液面(即玻璃管421的下端)与溶胀凝胶414的底面的高低差设为4cm。换言之,根据装置400,能够向盒411中导入规定静水压力的液体423。在活塞412中开通有孔415,因此,液体423在孔415中流通,进而也在溶胀凝胶414层中流通,并向盒411的外部流出。容器410载置在不妨碍液体423通过的不锈钢制的金属网431上。因此,从盒411中流出的液体423最终被捕集容器432收集。并且,被捕集容器432收集的液体423的量可利用上皿天平433进行称量。
[0082]
具体的凝胶渗透速度(gpr)的测定方法如下所示。需要说明的是,以下的操作在室温(20~25℃)下进行。
[0083]
(1)向盒411中均匀地投入颗粒状吸水剂(0.900g)。
[0084]
(2)使上述颗粒状吸水剂在未加压下吸收液体(0.90质量%的氯化钠水溶液)60分钟,制成溶胀凝胶414。
[0085]
(3)在溶胀凝胶414上载置活塞,制成0.3psi(2.06kpa)的加压状态。
[0086]
(4)边将静水压力保持为3923dyne/cm2的恒定值,边将液体423导入至盒411中,使溶胀凝胶414层进行通液。
[0087]
(5)以5秒钟的间隔对在溶胀凝胶414层中通液的液体423的量记录3分钟。即,测定在溶胀凝胶414层中通过的液体423的流速。在测定中使用上皿天平433和计算机(未图示)。
[0088]
(6)将自液体423开始流通起1分钟后~3分钟后的流速加以平均,算出凝胶渗透速度(gpr)[g/min]。
[0089]
本发明的颗粒状吸水剂的凝胶渗透速度(gpr)优选为20g/min以上。通过使凝胶渗透速度(gpr)为20g/min以上,从而将颗粒状吸水剂用于吸收体时的液体扩散性优异。颗粒状吸水剂的凝胶渗透速度(gpr)更优选为22g/min以上、进一步优选为24g/min以上、特别优选为26g/min以上。关于上限值,没有特别限定,优选为300g/min以下、更优选为150g/min以下。
[0090]
凝胶渗透速度(gpr)可通过控制例如吸水性树脂的内部交联剂量、表面交联剂量、表面交联反应的时间等来控制其数值。
[0091]
(2-4)吸湿结块率
[0092]
本发明中的“吸湿流动性”是针对将颗粒状吸水剂在气温为25℃且相对湿度为90%rh的条件下放置1小时时的结块、成饼或粉体形式的流动性进行评价的指标,可以用吸湿结块率进行判断。吸湿结块率的计算方法在实施例中详述。简而言之,将颗粒状吸水剂承载在筛上,进行分级,测定残留在筛上的颗粒状吸水剂的重量(w1[g])和通过了筛的颗粒状吸水剂的重量(w2[g]),按照下式来计算吸湿流动性。
[0093]
吸湿结块率[重量%]={w1/(w1 w2)}
×
100。
[0094]
测定方法的详情如实施例所述那样。
[0095]
本发明的颗粒状吸水剂的吸湿结块率通常为50重量%以下、优选为40重量%以下、更优选为30重量%以下、进一步优选为20重量%以下、更进一步优选为10重量%以下、最优选为0重量%。本发明的颗粒状吸水剂的吸湿结块率可以为0~50重量%、0~40重量%、0~30重量%、0~20重量%或0~10重量%。通过使吸湿结块率为40重量%以下,从而即便在多湿环境下,颗粒状吸水剂的处理性也良好,在制造面向卫生材料的薄型吸收体时等,出现在制造设备的输送配管内发生聚集和堵塞、无法与亲水性纤维均匀混合的问题的可能性变小。因此,通过满足上述条件,从而在使用颗粒状吸水剂和纤维基材来制作吸收体时,能够减少对装置设备的附着。
[0096]
吸湿结块率可通过为了提高吸湿时的流动性而添加的制剂的种类或其添加量来控制。
[0097]
(2-5)流下速度(flow rate、(ert450.2-02))
[0098]
流下速度(flow rate)是指颗粒状吸水剂的粉体流动性。
[0099]
具体而言,将颗粒状吸水剂100g投入至下部具备挡板的漏斗中,打开挡板,测量从开始流下至结束流下为止的时间,由此计算每单位时间的颗粒状吸水剂的流下量,将其作为流下速度。
[0100]
本发明的颗粒状吸水剂的流下速度(flow rate)优选为8.5g/s以上。流下速度小于8.5g/s时,颗粒状吸水剂的流动性低,因此,有时会产生如下问题:难以向料斗中供给颗粒状吸水剂、难以利用进料器来运输颗粒状吸水剂的问题;在制造面向卫生材料的吸收体时无法与亲水性纤维均匀混合的问题。
[0101]
(2-6)粉尘量
[0102]
本发明的颗粒状吸水剂的粉尘量相对于吸水剂优选为400mg/kg以下。通过使吸水剂的粉尘量为上述上限以下,从而充分降低粉尘,吸水剂的处理性优异。吸水剂的粉尘量相对于吸水剂更优选为300mg/kg以下、进一步优选为250mg/kg以下、特别优选为200mg/kg以下。该值为0mg/kg吸水剂是理想的,考虑到实际使用中和工业规模中的生产率,下限值相对于吸水剂通常为10mg/kg以上,可以为15mg/kg以上,也可以为20mg/kg以上。
[0103]
颗粒状吸水剂的粉尘量利用实施例所述的方法来计算。
[0104]
(2-7)表面张力
[0105]
表面张力是指以每单位面积的形式来表示对于增加固体、液体的表面积而言所需的功(自由能)。本技术中提及的表面张力是指:使颗粒状吸水剂分散在0.90质量%的氯化
钠水溶液中时的水溶液的表面张力。需要说明的是,吸水剂的表面张力按照以下的步骤来测定。即,向充分清洗的100ml烧杯中投入调整至20℃的生理盐水50ml,首先使用表面张力计(kruss公司制的k11自动表面张力计)来测定生理盐水的表面张力。接着,向包含调整至20℃且测定表面张力后的生理盐水的烧杯中投入充分清洗的25mm长的氟树脂制转子和颗粒状吸水剂0.5g,在500rpm的条件下搅拌4分钟。在4分钟后停止搅拌,在含水的颗粒状吸水剂发生沉降后,再次进行相同的操作来测定上清液的表面张力。需要说明的是,本发明中,采取使用白金板的板法,板在各测定之前充分用去离子水清洗,且利用气体燃烧器进行加热清洗来使用。
[0106]
本发明的颗粒状吸水剂的表面张力优选为65[mn/m]以上、更优选依次为66[mn/m]以上、68[mn/m]以上、70[mn/m]以上、71[mn/m]以上、72[mn/m]以上。通过使表面张力满足上述条件,从而能够进一步降低纸尿布中的回流量。上限通常为75[mn/m]就足够。
[0107]
(2-8)颗粒形状
[0108]
作为吸水性树脂(粉末)的颗粒形状,优选为不规则破碎状。此处,不规则破碎状是指形状不固定的破碎状颗粒。与通过反相悬浮聚合、气相聚合而得到的球状颗粒相比,不规则破碎状的形状不固定,因此,与纸浆等亲水性纤维的混合性优异,由颗粒间的间隙实现的液体扩散性高,故而优选。本发明的一个实施方式所述的颗粒状吸水剂优选为水溶液聚合中的粉碎物。不规则破碎状通过对经过水溶液聚合而得到的交联聚合物的凝胶或干燥物(优选为干燥物)进行粉碎来获得。另一方面,在未经过粉碎工序的情况下,代表而言,通过反相悬浮聚合、对聚合单体进行喷雾并聚合那样的液滴聚合等而得到的球状颗粒或球状颗粒的造粒物不是不规则破碎状。本发明的实施方式中,若颗粒状吸水剂的形状为不规则破碎状,则与平均圆度高的物质(例如球形物质)相比,吸水速度、rcap优异。本发明的实施方式中,颗粒状吸水剂的平均圆度优选为0.70以下、更优选为0.60以下、进一步优选为0.55以下。
[0109]
平均圆度的计算方法如下所示。随机地选择100个以上的颗粒状吸水剂,利用电子显微镜(基恩士公司制、ve-9800)(倍率为50倍)对各颗粒状吸水剂进行拍摄,获取颗粒状吸水剂的图像,使用附带的图像分析软件,计算每个颗粒的周长和面积。利用下式来求出各颗粒的圆度。
[0110]
[数学式2]
[0111]
圆度=4
×
π
×
面积/(周长)2[0112]
计算所得值的平均值来作为平均圆度。
[0113]
(2-9)水不溶性无机颗粒/水溶性的含有多价金属阳离子的化合物
[0114]
本发明的颗粒状吸水剂优选还包含选自由水不溶性无机颗粒和水溶性的含有多价金属阳离子的化合物组成的组中的至少1种。
[0115]
颗粒状吸水剂通过包含水不溶性无机颗粒而能够提高颗粒状吸水剂的吸湿流动性。另外,通过添加水不溶性无机颗粒而能够实现吸收性物品的吸收量的提高。进而,吸水性树脂颗粒(组合物)有时因制造后的保管而导致在制造吸收性物品时丧失流动性。通过对这种丧失了流动性的吸水性树脂颗粒(组合物)混合水不溶性无机颗粒,使其适合地成形为吸收体,从而在维持性能的同时,吸水性树脂颗粒(组合物)的流动性得以恢复,因此,生产率提高。此处,吸湿流动性是指在高湿条件下保管时的颗粒状吸水剂的流动性,包含吸水性
树脂的颗粒状吸水剂通常因吸湿而导致其流动性降低。需要说明的是,出于提高颗粒状吸水剂的吸湿流动性的目的,至今为止进行了水不溶性无机颗粒的添加,但单纯添加水不溶性无机颗粒并混合时,无法满足式(1)的关系,颗粒状吸水剂在溶胀状态下的加压下的回流量会增加(参照后述比较例)。另一方面,通过改进水不溶性无机颗粒的添加/混合条件(例如混合时间)等,从而能够使颗粒状吸水剂满足式(1)的关系,能够显著地抑制颗粒状吸水剂在溶胀状态下的加压下的液体回流。
[0116]
作为水不溶性无机颗粒,可列举出水滑石等多元金属化合物、二氧化硅(硅石)、氢氧化铝、二氧化钛、氧化铝、氧化镁、氧化锌、滑石、金属磷酸盐(例如磷酸三钙等磷酸钙、磷酸钡、磷酸铝)、金属硼酸盐(例如硼酸钛、硼酸铝、硼酸铁、硼酸镁、硼酸锰、硼酸钙)、硅酸或其盐、粘土、硅藻土、沸石、皂土、高岭土、活性白土等。其中,从显著获得本发明效果的方面出发,水不溶性无机颗粒优选包含选自多元金属化合物、二氧化硅、滑石和磷酸三钙中的至少1种,更优选包含选自二氧化硅、氢氧化铝和磷酸三钙中的至少1种。
[0117]
作为水不溶性无机颗粒的体积平均粒径,优选为10μm以下、更优选为5μm以下、进一步优选为1μm以下。另外,体积平均粒径优选为0.05μm以上、更优选为0.1μm以上、进一步优选为0.3μm以上。通过为上述下限以上,从而能够抑制添加工序时的作业性降低,得到充分的性能。需要说明的是,水不溶性无机颗粒的体积平均粒径可利用“激光衍射散射法”(例如使用日机装公司制、商品名:microtrac mt3000ii粒度分析计进行测定)来测定。
[0118]
水不溶性无机颗粒可以进行了表面处理。作为表面处理中使用的表面处理剂,可列举出下述多元金属化合物的表面处理剂的具体例。
[0119]
上述多元金属化合物是指含有2价和3价的两种金属阳离子且含有羟基的多元金属化合物。
[0120]
作为上述2价的金属阳离子,可列举出mg
2
、fe
2
、zn
2
、ca
2
、ni
2
、co
2
、cu
2
,从耐热性等观点出发,优选为mg
2
。作为上述3价的金属阳离子,可列举出al
3
、fe
3
、mn
3
,从耐热性等观点出发,优选为al
3
。因此,多元金属化合物的一个适合的实施方式是:2价的金属阳离子为镁阳离子,3价的金属阳离子为铝阳离子。
[0121]
多元金属化合物优选具有作为通式(1)[m
12 1-xm23 x
(oh-)2]
x
·
[(a
n-)
x/n
·
mh2o]
x-(m
12
表示2价的金属阳离子,m
23
表示3价的金属阳离子,a
n-表示n价的阴离子,h2o表示水)所示层状化合物的结构而已知的水滑石状结构。
[0122]
另外,关于通式(1)中的2价的金属阳离子与3价的金属阳离子的比率,x优选为0.2~0.75的范围,更优选为0.25~0.7的范围,进一步优选为0.25~0.5的范围。另外,作为阴离子,可列举出oh-、f-、cl-、br-、no
3-、co
32-、so
42-、fe(cn)
63-、ch3coo-、草酸根离子或水杨酸根离子等,优选为碳酸根阴离子。另外,m为大于0的实数,优选为0《m≤10。
[0123]
多元金属化合物的形状没有特别限定,优选为球状(包含粉末状)。另外,多元金属化合物优选为规定的粒度,体积平均粒径优选为2μm以下,更优选为1.5μm以下,进一步优选为1μm以下。通过使粒径为上述上限以下,从而用于获得充分效果的添加量不会过多,损害所得吸水剂的吸水性能的可能性小。另外,体积平均粒径优选为0.05μm以上,更优选为0.1μm以上,进一步优选为0.3μm以上。通过为上述下限以上,从而能够抑制添加工序时的作业性降低,能够得到充分的性能。另外,在吸水性树脂颗粒的表面附着的多元金属化合物的平均粒径的测定可通过使用sem(扫描型电子显微镜)的测定方法来进行测定。
[0124]
进而,可以向层间插入有机化合物,可以实施用于提高与吸水性树脂颗粒等的混合性的表面处理。
[0125]
作为多元金属化合物的优选结构式,可列举出mg6al2(oh)
16
co3·
4h2o、mg4al2(oh)
12
co3·
3h2o等。具体而言,可列举出协和化学工业公司制的dht-4h、dht-6、堺化学工业公司制的stabiace ht-1-nc、stabiace ht-p等。
[0126]
从提高吸收性物品的吸收量、制成吸收量与回流量的平衡优异的物品的观点出发,水不溶性无机颗粒的含量相对于聚丙烯酸(盐)系吸水性树脂100重量%为0.01重量%以上且小于10重量%,优选为0.1~5重量%。
[0127]
颗粒状吸水剂通过包含水溶性的含有多价金属阳离子的化合物而使吸水剂的性能提高。需要说明的是,出于提高颗粒状吸水剂的吸湿流动性的目的,至今为止进行了水溶性的含有多价金属阳离子的化合物的添加,但通过例如改进水溶性的含有多价金属阳离子的化合物的添加/混合条件(例如混合时间)等而不是单纯添加水溶性的含有多价金属阳离子的化合物并混合,从而能够满足式(1)的关系,能够得到显示出充分的加压下的吸水性能的颗粒状吸水剂。
[0128]
水溶性的含有多价金属阳离子的化合物是指除含有2价以上、优选3价以上的金属阳离子的多元金属化合物之外的化合物。作为该3价以上的金属阳离子,可例示出铝、锆、钛,优选为铝。作为该水溶性的含有多价金属阳离子的化合物,可列举出硫酸铝、氯化铝、氧氯化锆、碳酸锆铵、碳酸锆钾、碳酸锆钾、硫酸锆、乙酸锆、硝酸锆等多价金属的无机盐;乙酸铝、乳酸铝、羟基氯化锆、三乙醇胺酸钛(titanium triethanolaminate)、乳酸钛等多价金属的有机盐等多价金属化合物等。其中,优选为含有铝作为多价金属阳离子的化合物,更优选为硫酸铝、硫酸钾铝、硫酸钠铝。
[0129]
从提高吸水剂的性能的观点出发,水溶性的含有多价金属阳离子的化合物的含量相对于聚丙烯酸(盐)系吸水性树脂100重量份,换算成多价金属阳离子量优选为0.001~5重量份、更优选为0.01~2重量份、进一步优选为0.01~1重量份。
[0130]
上述形态的颗粒状吸水剂可通过例如在下述制造方法(a)~(c)中以满足式(1)的方式进行控制来获得。
[0131]
(制造方法a)
[0132]
向吸水性树脂中添加水不溶性无机颗粒,控制混合时间,得到满足式(1)的颗粒状吸水剂。
[0133]
要添加水不溶性无机颗粒的吸水性树脂可以进行了表面交联,也可以未经表面交联。另外,水不溶性无机颗粒可以与含有吸水性树脂或其它添加剂的吸水性树脂组合物进行混合。进而,在制造吸收性物品时,也可以将吸水性树脂(组合物)、水不溶性无机颗粒和亲水性纤维进行混合。
[0134]
另外,吸水性树脂与水不溶性无机颗粒优选进行干式混合。通过干式混合,所得吸水剂的粉尘量会降低,故而优选。该干式混合是指:在实质上不存在(优选不存在)除了水不溶性无机颗粒和吸水性树脂中吸收或保持的液状物质之外的液状物质的状态下的混合。具体而言,包括如下形态:在不进一步添加液状物质的条件下,将包含吸湿水分、在层之间保持的有机化合物的水不溶性无机颗粒与具有干燥残量、吸湿水分、在前述表面交联剂添加工序中添加的表面交联剂、溶剂等的吸水性树脂进行混合的形态。
[0135]
吸水性树脂与水不溶性无机颗粒的混合时间没有特别限定,可利用混合装置来适当设定,优选以满足上述式(1)的方式进行较长时间的混合。即,在本发明中,优选设定为比出于单纯混合添加剂的目的时的混合时间更长的时间,将吸水性树脂与水不溶性无机颗粒加以混合。通常,出于提高生产率的目的,在可通过目视或以往的评价方法而确认到添加物已经均匀混合的时刻,停止混合装置的运行。另外,通过这种以往的混合,也确保了aap、crc之类的对颗粒状吸水剂要求的基本性能。然而,这些颗粒状吸水剂在颗粒状吸水剂发生溶胀的状态下的加压下的回流量并未充分降低。本发明人等发现这种课题并进行了研究,其结果作出如下假设:可能是因为水不溶性无机颗粒容易聚集,因此,其在吸水性树脂中未被微观性地充分分散,在添加水不溶性无机颗粒时,会导致颗粒状吸水剂的物性降低、尤其是溶胀状态下的加压下的回流量增加。并且发现:通过使混合水不溶性无机颗粒时的混合时间长于对于通常的均匀混合而言所需的时间,从而能够得到用于满足式(1)的颗粒状吸水剂。一般来说,均匀混合所需的时间通过以往的评价方法等来设定,但本发明人等发现了rcap这一新型参数成为用于改善在溶胀状态下的加压下的回流量的一个指标,由此,首先发现通过进一步延长以往被视作是充分的混合时间而实现的附加效果。以往认为,较长的混合时间会导致生产率的降低等,反而是不优选的,因此,并未积极地加以实施,但在本发明中,根据上述发现,能够得到远超生产率降低等缺点的更大的优点。吸水性树脂与水不溶性无机颗粒的混合时间例如为5分钟以上,优选为15分钟以上,更优选为30分钟以上。
[0136]
在吸水性树脂与水不溶性无机颗粒的混合中使用的手段没有特别限定,优选使用成为较强搅拌条件那样的混合手段,而不是桨式搅拌装置那样的温和搅拌。
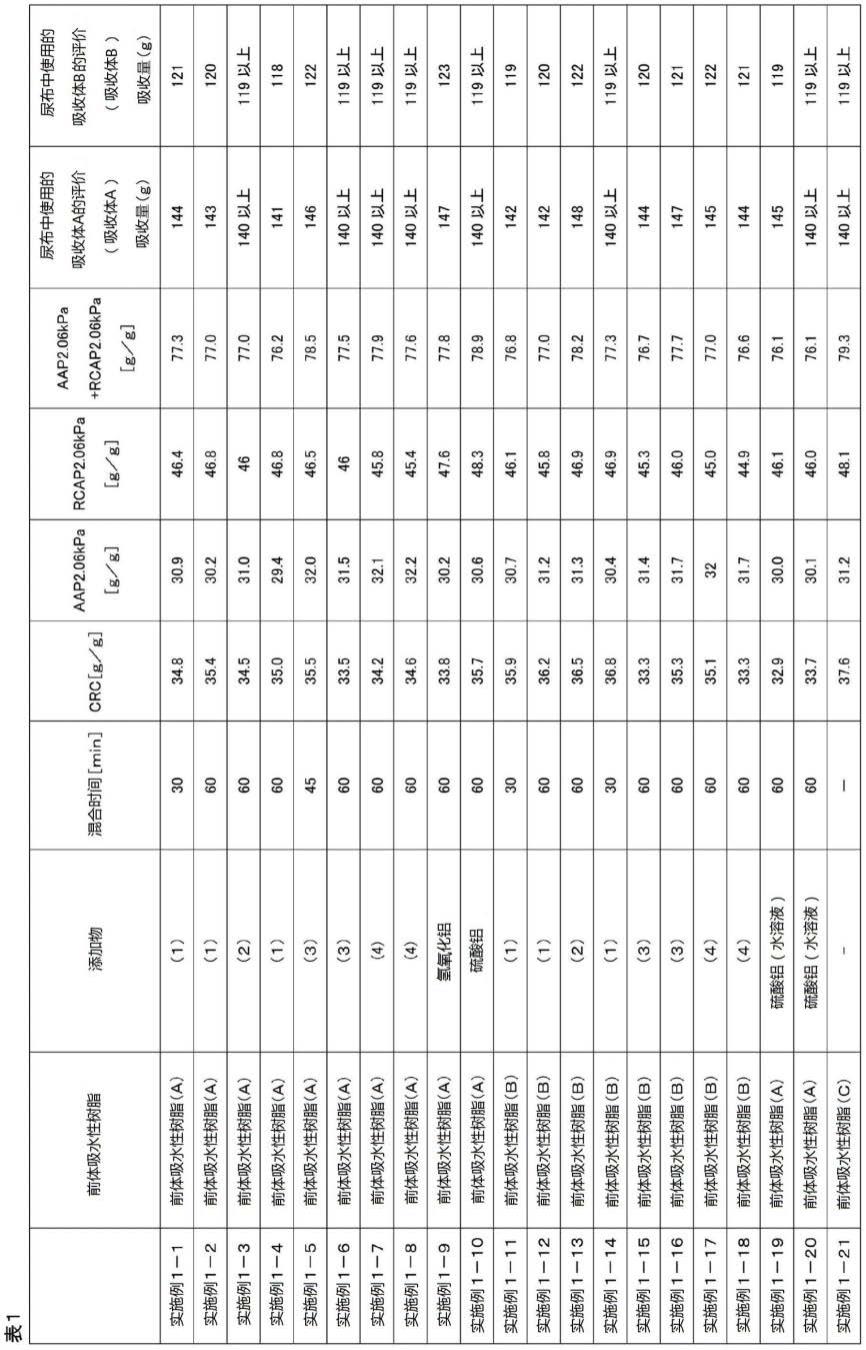
[0137]
混合时间的上限没有特别限定,若考虑到生产率、效果的饱和,则优选为5小时以下、更优选为2小时以下。另外,作为混合方法,没有特别限定,优选为边施加振动边混合的方法、边旋转运动边混合的方法(例如使用转筒摇摆混合机的方法)、基于搅拌机的混合、空气输送等与气流一同运输颗粒那样的方法。搅拌条件根据混合装置来适当设定,例如,在使用转筒摇摆混合机的情况下,其旋转速度优选高于45rpm,更优选为70rpm以上,进一步优选为100rpm以上。另外,对于理想的混合而言所需的混合时间因各种混合方法而异,通过适当调整混合时间而能够实现本技术的目的。关于能否实现本技术目的的判断,是否满足本技术的物性参数可成为一个指标。
[0138]
(制造方法b)
[0139]
向吸水性树脂中添加水溶性的含有多价金属阳离子的化合物,控制混合时间,得到满足式(1)的颗粒状吸水剂。
[0140]
水溶性的含有多价金属阳离子的化合物可以以粉体的形式直接混合于吸水性树脂,也可以制成溶液、尤其是水溶液来进行混合,还可以溶解于表面交联剂或其水溶液来混合。
[0141]
另外,可以多次添加,在该情况下,例如添加2次时,作为其(重量)比率,规定为1/99~99/1、优选规定为10/90~90/10的范围。若超过这些范围,则极其近似于与添加1次相同的状况,缺乏多次添加的效果,故不优选。
[0142]
作为水溶性的含有多价金属阳离子的化合物的添加方法,没有特别限定,优选的是:(1)在添加表面交联剂的同时进行添加(在表面交联工序中进行添加);(2)在表面交联工序之后进行添加。通过在这种时刻添加水溶性的含有多价金属阳离子的化合物,从而能
够存在于颗粒表面附近,能够提高吸水剂的性能。在表面交联工序之后进行添加时,也可以与其它添加剂一同添加。
[0143]
水溶性的含有多价金属阳离子的化合物可以在溶解于溶剂(例如水)的溶液状态下添加于吸水性树脂(粉末)。以水溶液的形式添加水溶性的含有多价金属阳离子的化合物时,除了水之外,也可以组合使用亲水性有机溶剂(醇或者聚二醇)、表面活性剂来提高分散性、溶解性、混合性。所使用的水量根据水溶性的含有多价金属阳离子的化合物的种类、添加方法来适当决定,例如,相对于吸水性树脂100重量份为0重量份(干式混合)~50重量份,进而为0.1~10重量份、0.5~5重量份。
[0144]
另外,水溶性的含有多价金属阳离子的化合物可以在其原有形态下添加于吸水性树脂(粉末)。在该情况下,水溶性的含有多价金属阳离子的化合物优选为颗粒形态。作为水溶性的含有多价金属阳离子的化合物颗粒的体积平均粒径,优选为10μm以下、更优选为5μm以下、进一步优选为1μm以下。另外,体积平均粒径优选为0.05μm以上、更优选为0.1μm以上、进一步优选为0.3μm以上。通过为上述下限以上,从而能够抑制添加工序时的作业性降低,能够得到充分的性能。需要说明的是,水溶性的含有多价金属阳离子的化合物颗粒的体积平均粒径可利用“激光衍射散射法”(例如,使用日机装公司制、商品名:microtrac mt3000ii粒度分析计进行测定)来测定。
[0145]
本说明书中,水溶性表示在室温(23℃)、常压下(1个大气压下)可溶于(或易溶于)水的物质。例如,表示在室温、常压下相对于水100ml而言的溶解量为1g以上的物质。另外,水不溶性表示在室温(23℃)、常压下(1个大气压下)不溶于(或难溶于)水的物质。例如,表示在室温、常压下相对于水100ml而言的溶解量小于1g的物质,进一步优选在室温、常压下相对于水100ml而言的溶解量小于0.1g。
[0146]
吸水性树脂与水溶性的含有多价金属阳离子的化合物的混合时间没有特别限定,可利用混合装置来适当设定,优选以满足上述式(1)的方式进行较长时间的混合。即,在本发明中,优选设定为比出于单纯混合添加剂的目的时的混合时间更长的时间,将吸水性树脂与水溶性的含有多价金属阳离子的化合物进行混合。通常,出于提高生产率的目的,在可通过目视或以往的评价方法而确认到添加物已经均匀混合的时刻,停止混合装置的运行。另外,通过这种以往的混合,也确保了aap、crc之类的对颗粒状吸水剂要求的基本性能。然而,这些颗粒状吸水剂在颗粒状吸水剂发生溶胀的状态下的加压下的回流量并未充分降低。本发明人等发现这种课题并进行了研究,其结果作出如下假设:可能是因为水溶性的含有多价金属阳离子的化合物在以往的混合方法和时间中是不充分的,因此,其在吸水性树脂中未被微观性地充分分散,在添加水溶性的含有多价金属阳离子的化合物时,会导致颗粒状吸水剂的物性降低、尤其是溶胀状态下的加压下的回流量增加。并且发现:通过使混合水溶性的含有多价金属阳离子的化合物时的混合时间长于对于通常的均匀混合而言所需的时间,从而能够得到用于满足式(1)的颗粒状吸水剂。一般来说,均匀混合所需的时间通过以往的评价方法等来设定,但本发明人等发现了rcap这一新型参数成为用于改善在溶胀状态下的加压下的回流量的一个指标,由此,首先发现通过进一步延长以往被视作是充分的混合时间而实现的附加效果。以往认为:较长的混合时间会导致生产率的降低等,反而是不优选的,因此,并未积极地加以实施,但在本发明中,根据上述发现,能够得到远超生产率降低等缺点的更大的优点。吸水性树脂与水溶性的含有多价金属阳离子的化合物的混合时
间例如为5分钟以上,优选为15分钟以上,更优选为30分钟以上。混合时间的上限没有特别限定,若考虑到生产率、效果的饱和,则优选为5小时以下、更优选为2小时以下。另外,作为混合方法,没有特别限定,优选为边施加振动边混合的方法、边旋转运动边混合的方法(例如使用转筒摇摆混合机的方法)、基于搅拌机的混合、空气输送等与气流一同输送颗粒那样的方法。此时的转速也可以与制造方法a相同。另外,对于理想的混合而言所需的混合时间因各种混合方法而异,通过适当调整混合时间而能够实现本技术的目的。关于能否实现本技术目的的判断,是否满足本技术的物性参数可成为一个指标。
[0147]
(制造方法c)
[0148]
在制造颗粒状吸水剂的制造方法中,具有用包含水作为主成分的清洗液来清洗吸水性树脂的工序。通过利用以水作为主成分的清洗液来清洗吸水性树脂,从而容易得到满足式(1)的颗粒状吸水剂。详细机理尚不明明确,可以认为:通过利用包含水作为主成分的清洗液来清洗吸水性树脂,从而吸水性树脂含有水而呈现溶胀状态,通过在该状态下进行清洗,从而能够有效地洗掉可能影响吸水性能的无用成分,容易得到rcap高的吸水剂。
[0149]
需要说明的是,在上述专利文献2(国际公开第97/003114号)中,出于降低残留交联剂的目的而进行了清洗,“以混合液不使吸水性树脂粉末发生溶胀的方式来选择水与亲水性有机溶剂的混合比范围”,因此,与通过利用包含水作为主成分的清洗液进行清洗而使吸水性树脂粉末发生溶胀的上述工序明显不同。
[0150]
作为清洗方法,优选的是:例如在将吸水性树脂制成溶胀状态后,使用清洗液进行清洗。作为将吸水性树脂制成溶胀状态的方法,可列举出:使用以水作为主成分的液体,将吸水性树脂浸渍于该液体的方法。作为浸渍时间,只要吸水性树脂充分发生溶胀,就没有特别限定,例如,可以为1分钟以上,也可以为5分钟以上。
[0151]
水为主成分是指:在清洗液中,包含80重量%以上、优选90重量%以上、更优选95重量%以上、更进一步优选99重量%以上的水,优选实质上由水组成。
[0152]
水优选不含杂质,优选为ro水、去离子水、蒸馏水、纯化水等,更优选为去离子水、蒸馏水。
[0153]
作为除水之外的成分,可以包含亲水性有机溶剂。作为所使用的亲水性有机溶剂,可适当地例示出甲醇、乙醇、丙醇、异丙醇、叔丁醇等低级醇类。
[0154]
此处,吸水性树脂可以为表面交联处理前的吸水性树脂粉末、表面交联后的吸水性树脂颗粒中的任一者,从更容易得到满足式(1)的颗粒状吸水剂的方面出发,优选对表面交联处理前的吸水性树脂粉末(所谓的基础聚合物)进行清洗。
[0155]
作为清洗方法,没有特别限定,可以利用清洗液和连续或不连续的间歇式方法对吸水性树脂进行清洗。可列举出例如:使吸水性树脂在清洗液中根据需要边搅拌边进行接触,其后,利用例如倾析、抽滤而从清洗液中分离吸水性树脂的方法;对溶胀状态的含水凝胶通入清洗液的通水清洗等。
[0156]
另外,清洗也可以进行多次。
[0157]
在这些方法中,清洗时间(通水时间)优选为15分钟~10小时,更优选为30分钟~8小时,进一步优选为1~5小时。从清洗效果的方面出发,清洗液的温度优选为20~50℃。另外,清洗时的压力为加压、减压、常压均可,通常在常压下进行。
[0158]
清洗后的吸水性树脂(含水凝胶)与下述〔3〕颗粒状吸水剂的制造方法中的含水凝
胶的后续工序同样,根据需要进一步追加进行干燥、粉碎、分级等工序即可。
[0159]
〔3〕颗粒状吸水剂的制造方法
[0160]
以下,示出本发明涉及的颗粒状吸水剂的制造工序(3-1)~(3-8)。
[0161]
(3-1)单体水溶液的制备工序
[0162]
本工序是制备包含单体(例如丙烯酸(盐))作为主成分的水溶液(以下称为“单体水溶液”)的工序。需要说明的是,可以在所得吸水性树脂的吸水性能不会降低的范围内使用单体的浆料液,但在本项中,为了方便,针对单体水溶液进行说明。
[0163]
另外,上述“主成分”是指:丙烯酸(盐)的用量(含量)相对于供于吸水性树脂的聚合反应的单体(不包括内部交联剂)整体而言,通常为50摩尔%以上、优选为70摩尔%以上、更优选为90摩尔%以上(上限为100摩尔%)。
[0164]
(丙烯酸)
[0165]
本发明中,从所得颗粒状吸水剂的物性和生产率的观点出发,作为单体,使用丙烯酸和/或其盐(以下称为“丙烯酸(盐)”)。
[0166]
上述“丙烯酸”可以是公知的丙烯酸,作为阻聚剂,优选包含甲氧基苯酚类、更优选包含对甲氧基苯酚,从丙烯酸的聚合性、颗粒状吸水剂的色调的观点出发,优选包含200ppm以下、更优选包含10~160ppm、进一步优选包含20~100ppm即可。另外,关于丙烯酸中的杂质,在本发明中也可应用美国专利申请公开第2008/0161512号所记载的化合物。
[0167]
另外,上述“丙烯酸盐”是将上述丙烯酸用下述碱性组合物进行中和而得到的物质,作为该丙烯酸盐,可以为市售的丙烯酸盐(例如丙烯酸钠),也可以为在颗粒状吸水剂的制造设备内进行中和而得到的物质。
[0168]
(碱性组合物)
[0169]
本发明中,“碱性组合物”是指含有碱性化合物的组合物,例如市售的氢氧化钠水溶液等是符合的。
[0170]
作为上述碱性化合物,具体而言,可列举出碱金属的碳酸盐、碳酸氢盐、碱金属的氢氧化物、氨、有机胺等。这些之中,从所得颗粒状吸水剂的物性的观点出发,期望为强碱性。即,优选为氢氧化钠、氢氧化钾、氢氧化锂等碱金属的氢氧化物,更优选为氢氧化钠。
[0171]
(中和)
[0172]
作为本发明中的中和,可以选择或组合使用对丙烯酸进行的中和(聚合前)或者对将丙烯酸交联聚合而得到的含水凝胶状交联聚合物进行的中和(聚合后)(以下称为“后中和”)中的任意者。另外,这些中和可以为连续式,也可以为间歇式,没有特别限定,从生产效率等的观点出发,优选为连续式。
[0173]
需要说明的是,关于进行中和的装置、中和温度、滞留时间等条件,也可以在本发明中应用国际公开第2009/123197号、美国专利申请公开第2008/0194863号所记载的条件。
[0174]
本发明中的中和率相对于单体的酸基优选为10~90摩尔%、更优选为40~85摩尔%、进一步优选为50~80摩尔%、特别优选为60~75摩尔%。该中和率小于10摩尔%时,吸水倍率有时显著降低。另一方面,该中和率超过90摩尔%时,有时得不到加压下的吸水倍率高的吸水性树脂。
[0175]
上述中和率在后中和的情况下也同样。另外,关于作为最终制品的颗粒状吸水剂的中和率,也可应用上述中和率。需要说明的是,中和率为75摩尔%是指丙烯酸25摩尔%与
丙烯酸盐75摩尔%的混合物。另外,有时也将该混合物称为丙烯酸部分中和物。
[0176]
(其它单体)
[0177]
在本发明中,“其它单体”是指除上述丙烯酸(盐)之外的单体,可以将其它单体与丙烯酸(盐)组合使用来制造颗粒状吸水剂。
[0178]
作为上述其它单体,可列举出水溶性或疏水性的不饱和单体。具体而言,也可以在本发明中应用美国专利申请公开第2005/0215734所记载的化合物(其中不包括丙烯酸)。
[0179]
(内部交联剂)
[0180]
作为本发明中使用的内部交联剂,也可以在本发明中应用美国专利第6241928号所记载的化合物。可以考虑反应性而从这些之中选择1种或2种以上的化合物。本发明中,考虑到吸水性能,优选对使用了内部交联剂的交联体进行表面处理。
[0181]
另外,从所得吸水性树脂的吸水性能等观点出发,作为内部交联剂,优选使用具有2个以上聚合性不饱和基团的化合物,更优选使用在下述干燥温度下具有热分解性的化合物,进一步优选使用具有2个以上具有(聚)亚烷基二醇结构单元的聚合性不饱和基团的化合物。
[0182]
作为上述聚合性不饱和基团,可优选列举出烯丙基、(甲基)丙烯酸酯基,可更优选列举出(甲基)丙烯酸酯基。另外,作为上述(聚)亚烷基二醇结构单元,优选为聚乙二醇,作为n数,优选为1~100、更优选为6~50。
[0183]
因此,本发明中,优选使用(聚)亚烷基二醇二(甲基)丙烯酸酯或(聚)亚烷基二醇三(甲基)丙烯酸酯,更优选使用(聚)乙二醇二(甲基)丙烯酸酯。
[0184]
上述内部交联剂的用量相对于单体整体优选为0.0001~10摩尔%、更优选为0.001~1摩尔%。通过将该用量设为上述范围内,从而能够得到期望的吸水性树脂。需要说明的是,该用量过少时,存在凝胶强度降低、水可溶成分增加的倾向,该用量过多时,存在吸水倍率降低的倾向。
[0185]
本发明中,优选应用预先向单体水溶液中添加规定量的内部交联剂,并在聚合的同时进行交联反应的方法。另一方面,除了该方法之外,也可以采用如下方法:在聚合中、聚合后添加内部交联剂来进行后交联的方法;使用自由基聚合引发剂来进行自由基交联的方法;使用电子射线、紫外线等活性能量射线来进行辐射线交联的方法等。另外,还可以组合使用这些方法。
[0186]
(其它向单体水溶液中添加的物质)
[0187]
本发明中,从提高所得吸水性树脂的物性的观点出发,也可以在制备单体水溶液时添加下述物质。
[0188]
具体而言,可以向单体水溶液中添加优选为50重量%以下、更优选为20重量%以下、进一步优选为10重量%以下、特别优选为5重量%以下(下限为0重量%)的淀粉、淀粉衍生物、纤维素、纤维素衍生物、聚乙烯醇、聚丙烯酸(盐)、聚丙烯酸(盐)交联体等亲水性高分子,也可以向单体水溶液中添加优选为5重量%以下、更优选为1重量%以下、进一步优选为0.5重量%以下(下限为0重量%)的碳酸盐、偶氮化合物、气泡等发泡剂、表面活性剂、二乙烯三胺五乙酸(盐)、乙二胺四亚甲基膦酸(盐)等螯合剂、链转移剂等。
[0189]
另外,上述物质不仅可以是向单体水溶液中添加的形态,也可以是在聚合过程中进行添加的形态,还可以组合使用这些形态。
[0190]
需要说明的是,作为亲水性高分子而使用水溶性树脂或吸水性树脂时,能够得到接枝聚合物或吸水性树脂组合物(例如,淀粉-丙烯酸聚合物、pva-丙烯酸聚合物等)。这些聚合物、吸水性树脂组合物也属于本发明的范畴。
[0191]
(单体成分的浓度)
[0192]
在本工序中,在制备单体水溶液时,添加上述的各种物质。作为该单体水溶液中的单体成分的浓度,没有特别限定,从吸水性树脂的物性的观点出发,优选为10~80重量%、更优选为20~75重量%、进一步优选为30~70重量%。
[0193]
另外,采用水溶液聚合或反相悬浮聚合时,也可以根据需要而组合使用除水之外的溶剂。该情况下,溶剂的种类没有特别限定。
[0194]
需要说明的是,上述“单体成分的浓度”是指利用下述式(5)而求出的值,单体水溶液的重量中不包括接枝成分、吸水性树脂、反相悬浮聚合中的疏水性溶剂的重量。
[0195]
(单体成分的浓度(重量%))=(单体成分的重量)/(单体水溶液的重量)
×
100式(5)
[0196]
(3-2)聚合工序
[0197]
本工序是使通过上述单体水溶液的制备工序而得到的丙烯酸(盐)系单体水溶液进行聚合,从而得到含水凝胶状交联聚合物(以下称为“含水凝胶”)的工序。
[0198]
(聚合引发剂)
[0199]
本发明中使用的聚合引发剂根据聚合形态等来适当选择,因此没有特别限定,可列举出例如热分解型聚合引发剂、光分解型聚合引发剂、或者组合使用会促进这些聚合引发剂的分解的还原剂而得到的氧化还原系聚合引发剂等。具体而言,可以使用美国专利第7265190号所公开的聚合引发剂之中的1种或2种以上。需要说明的是,从聚合引发剂的处理性、颗粒状吸水剂或吸水性树脂的物性的观点出发,优选使用过氧化物或偶氮化合物,更优选使用过氧化物,进一步优选使用过硫酸盐。
[0200]
该聚合引发剂的用量相对于单体优选为0.001~1摩尔%、更优选为0.001~0.5摩尔%。另外,该还原剂的用量相对于单体优选为0.0001~0.02摩尔%。
[0201]
需要说明的是,代替上述聚合引发剂,可以照射辐射线、电子射线、紫外线等活性能量射线来实施聚合反应,也可以将这些活性能量射线与聚合引发剂进行组合使用。
[0202]
(聚合形态)
[0203]
作为本发明中应用的聚合形态,没有特别限定,从吸水特性、聚合控制容易性等观点出发,可优选列举出喷雾液滴聚合、水溶液聚合、反相悬浮聚合,可更优选列举出水溶液聚合、反相悬浮聚合,可进一步优选列举出水溶液聚合。其中,特别优选为连续水溶液聚合,连续带聚合、连续捏合机聚合均可应用。
[0204]
作为具体的聚合形态,分别在美国专利第4893999号、美国专利第6241928号、美国专利申请公开第2005/215734号等中公开了连续带聚合,在美国专利第6987151号、美国专利第6710141号等中公开了连续捏合机聚合。通过采用这些连续水溶液聚合,从而吸水性树脂的生产效率提高。
[0205]
另外,作为上述连续水溶液聚合的优选形态,可列举出“高温引发聚合”、“高浓度聚合”。“高温引发聚合”是指:在单体水溶液的温度优选为30℃以上、更优选为35℃以上、进一步优选为40℃以上、特别优选为50℃以上(上限为沸点)的温度下引发聚合的形态,“高浓
度聚合”是指:在单体浓度优选为30重量%以上、更优选为35重量%以上、进一步优选为40重量%以上、特别优选为45重量%以上(上限为饱和浓度)的条件下进行聚合的形态。也可以组合使用这些聚合形态。
[0206]
另外,本发明中,也可以在空气气氛下进行聚合,但从所得吸水性树脂的色调的观点出发,可以在氮气、氩气等非活性气体的气氛下进行聚合。在该情况下,例如,优选将氧浓度控制为1容积%以下。需要说明的是,关于单体水溶液中的溶解氧,也优选预先用非活性气体进行置换(例如溶解氧:小于1mg/l)。
[0207]
另外,在本发明中,也可以设为使气泡(尤其是上述非活性气体等)分散于单体水溶液而进行聚合的发泡聚合。
[0208]
另外,在本发明中,可以在聚合中使固体成分浓度上升。作为这种固体成分浓度上升的指标,固体成分上升度利用下述式(6)来定义。需要说明的是,作为该固体成分浓度的上升度,优选为1重量%以上、更优选为2重量%以上。(固体成分上升度(重量%))=(聚合后的含水凝胶的固体成分浓度(重量%))-(单体水溶液的固体成分浓度(重量%))式(6)。
[0209]
其中,单体水溶液的固体成分浓度是指利用下述式(7)而求出的值,聚合体系内的成分是指单体水溶液和接枝成分、吸水性树脂、其它固态物(例如水不溶性微粒等),不包括反相悬浮聚合中的疏水性溶剂。
[0210]
(单体水溶液的固体成分(重量%))=((单体成分 接枝成分 吸水性树脂 其它固态物)的重量)/(聚合体系内的成分的重量)
×
100式(7)。
[0211]
另外,作为水溶液聚合的形态,可利用在静置状态下将单体水溶液进行聚合的静置聚合法、在搅拌装置内进行聚合的搅拌聚合法等来实施本发明。在静置聚合法中,优选使用环状带。带优选为聚合热不易从材料接触面逸散的树脂或橡胶制的带。
[0212]
(3-3)凝胶粉碎工序
[0213]
本工序是利用例如捏合机、碎肉机等螺杆挤出机、切割磨等凝胶粉碎机对上述聚合工序中得到的含水凝胶进行凝胶粉碎,得到颗粒状的含水凝胶(以下称为“颗粒状含水凝胶”)的工序。需要说明的是,上述聚合工序为捏合机聚合时,可同时实施聚合工序和凝胶粉碎工序。另外,在气相聚合、反相悬浮聚合等在聚合过程中直接获得颗粒状含水凝胶的情况下,有时也不实施该凝胶粉碎工序。
[0214]
关于除上述之外的凝胶粉碎条件、形态,在本发明中优选应用国际公开第2011/126079号所公开的内容。
[0215]
(3-4)干燥工序
[0216]
本工序是使通过上述聚合工序和/或凝胶粉碎工序而得到的颗粒状含水凝胶干燥至期望的树脂固体成分而得到干燥聚合物的工序。该树脂固体成分根据干燥减量(将吸水性树脂1g以180℃加热3小时时的重量变化)来求出,优选为80重量%以上、更优选为85~99重量%、进一步优选为90~98重量%、特别优选为92~97重量%。
[0217]
作为上述颗粒状含水凝胶的干燥方法,没有特别限定,可列举出例如加热干燥、热风干燥、减压干燥、流动层干燥、红外线干燥、微波干燥、转筒干燥器干燥、基于与疏水性有机溶剂的共沸脱水而进行的干燥、利用高温水蒸气而进行的高湿干燥等。其中,从干燥效率的观点出发,优选为热风干燥,更优选为在通气带上进行热风干燥的带式干燥。
[0218]
作为上述热风干燥中的干燥温度(热风温度),从吸水性树脂的色调、干燥效率的
观点出发,优选为120~250℃、更优选为150~200℃。需要说明的是,关于热风风速、干燥时间等除了上述干燥温度之外的干燥条件,根据供于干燥的颗粒状含水凝胶的含水率、总重量和作为目标的树脂固体成分来适当设定即可,进行带式干燥时,可适当应用国际公开第2006/100300号、国际公开第2011/025012号、国际公开第2011/025013号、国际公开第2011/111657号等中记载的各条件。
[0219]
(3-5)粉碎工序、分级工序
[0220]
本工序是对通过上述干燥工序而得到的干燥聚合物进行粉碎(粉碎工序),调整至规定范围的粒度(分级工序),得到吸水性树脂粉末(将实施表面交联之前的粉末状的吸水性树脂简称为“吸水性树脂粉末”)的工序。
[0221]
作为本发明的粉碎工序中使用的机器,可列举出例如辊磨机、锤磨机、螺杆磨、销磨机等高速旋转式粉碎机;振动磨、转向节(knuckle)类型的粉碎机、圆筒型搅拌器等,可根据需要而组合使用。
[0222]
另外,作为本发明的分级工序中的粒度调整方法,没有特别限定,可列举出例如使用jis标准筛(jis z8801-1(2000))进行的筛分级、气流分级等。需要说明的是,吸水性树脂的粒度调整不限定于上述粉碎工序、分级工序,可利用聚合工序(尤其是反相悬浮聚合、喷雾液滴聚合)、其它工序(例如造粒工序、微粉回收工序)来适当实施。
[0223]
关于上述工序中得到的吸水性树脂粉末(表面交联工序前的吸水性树脂粉末、所谓的基础聚合物),作为重均粒径(d50),优选为200~600μm、更优选为200~550μm、进一步优选为250~500μm。另外,粒径小于150μm的颗粒的比例优选为10重量%以下、更优选为5重量%以下、进一步优选为1重量%以下,粒径为850μm以上的颗粒的比例优选为5重量%以下、更优选为3重量%以下、进一步优选为1重量%以下。需要说明的是,作为这些颗粒的比例的下限值,在任意情况下均是越少越优选,期望为0重量%,可以为0.1重量%左右。进而,粒度分布的对数标准偏差(σζ)优选为0.20~0.50、更优选为0.25~0.40、进一步优选为0.27~0.35。需要说明的是,这些粒度可按照美国专利第7638570号、edana ert420.2-02中公开的测定方法,使用标准筛来进行测定。
[0224]
上述粒度不仅可应用于表面交联后的吸水性树脂(以下有时简称为“吸水性树脂颗粒”),还可应用于作为最终制品的颗粒状吸水剂。因此,优选以在吸水性树脂颗粒中维持上述范围的粒度的方式进行表面交联处理(表面交联工序),更优选在表面交联工序中及之后设置整粒工序来进行粒度调整。
[0225]
(3-6)表面交联工序
[0226]
本工序是在经过上述工序而得到的吸水性树脂粉末的表面层(距离吸水性树脂粉末的表面为数10μm的部分)进一步设置交联密度高的部分的工序,由混合工序、加热处理工序和冷却工序(任选)构成。
[0227]
在该表面交联工序中,通过吸水性树脂粉末表面处的自由基交联、表面聚合、与表面交联剂发生的交联反应等,从而得到经表面交联的吸水性树脂(吸水性树脂颗粒)。
[0228]
(表面交联剂)
[0229]
作为本发明中使用的表面交联剂,没有特别限定,可列举出有机或无机的表面交联剂。其中,从吸水性树脂的物性、表面交联剂的处理性的观点出发,优选为与羧基发生反应的有机表面交联剂。可列举出例如美国专利7183456号中公开的1种或2种以上的表面交
联剂。更具体而言,可列举出多元醇化合物、环氧化合物、卤代环氧化合物、多胺化合物或其与卤代环氧化合物的缩合物、噁唑啉化合物、噁唑烷酮化合物、多价金属盐、碳酸亚烷基酯化合物、环状脲化合物等。
[0230]
作为有机表面交联剂的具体例,可列举出(二、三、四、聚)乙二醇、(二、聚)丙二醇、1,3-丙二醇、2,2,4-三甲基-1,3-戊二醇、(聚)甘油、2-丁烯-1,4-二醇、1,4-丁二醇、1,3-丁二醇、1,5-戊二醇、1,6-己二醇、三羟甲基丙烷、二乙醇胺或三乙醇胺、季戊四醇、山梨糖醇等多元醇化合物;(聚)乙二醇二缩水甘油醚、(二、聚)甘油聚缩水甘油醚、缩水甘油等环氧化合物;2-噁唑烷酮、n-羟基乙基-2-噁唑烷酮、1,2-乙烯双噁唑啉等噁唑啉化合物;1,3-二氧戊环-2-酮(碳酸亚乙酯)、4-甲基-1,3-二氧戊环-2-酮、4,5-二甲基-1,3-二氧戊环-2-酮、4,4-二甲基-1,3-二氧戊环-2-酮、4-乙基-1,3-二氧戊环-2-酮、4-羟基甲基-1,3-二氧戊环-2-酮、1,3-二噁烷-2-酮、4-甲基-1,3-二噁烷-2-酮、4,6-二甲基-1,3-二噁烷-2-酮、1,3-二氧杂环庚烷(dioxepane)-2-酮等碳酸亚烷基酯化合物;表氯醇、表溴醇、α-甲基表氯醇等卤代环氧化合物、及其多胺加成物(例如hercules公司制的kymene;注册商标);γ-环氧丙氧基丙基三甲氧基硅烷、γ-氨基丙基三乙氧基硅烷等硅烷偶联剂;3-甲基-3-氧杂环丁烷甲醇、3-乙基-3-氧杂环丁烷甲醇、3-丁基-3-氧杂环丁烷甲醇、3-甲基-3-氧杂环丁烷乙醇、3-乙基-3-氧杂环丁烷乙醇、3-丁基-3-氧杂环丁烷乙醇、3-氯甲基-3-甲基氧杂环丁烷、3-氯甲基-3-乙基氧杂环丁烷、多价氧杂环丁烷化合物等氧杂环丁烷化合物;2-咪唑烷酮等环状脲化合物等。
[0231]
作为前述多元醇,优选碳原子数为2~8的多元醇,更优选碳原子数为3~6的多元醇,进一步优选碳原子数为3~4的多元醇。进而,优选为二醇,可例示出乙二醇、丙二醇、1,3-丙二醇、1,4-丁二醇,优选为选自丙二醇(1,2-丙二醇)、1,3-丙二醇、1,4-丁二醇中的多元醇。
[0232]
另外,作为环氧化合物,优选为聚缩水甘油基化合物,适合使用乙二醇二缩水甘油醚。
[0233]
在上述有机表面交联剂的基础上,从更有效地进行表面交联的观点出发,作为离子键合性表面交联剂,可组合使用多胺聚合物等多价阳离子性聚合物、水溶性的含有多价金属阳离子的化合物。作为水溶性的含有多价金属阳离子的化合物,如上所述。
[0234]
该表面交联剂的用量(使用多种时是总用量)相对于吸水性树脂粉末100重量份优选为0.01~10重量份、更优选为0.01~5重量份。另外,该表面交联剂优选以水溶液的形式添加,在该情况下,水的用量相对于吸水性树脂粉末100重量份优选为0.1~20重量份、更优选为0.5~10重量份。进而,根据需要而使用亲水性有机溶剂时,其用量相对于吸水性树脂粉末100重量份优选为10重量份以下、更优选为5重量份以下。
[0235]
另外,如上所述那样,在该表面交联工序中,可以添加水溶性的含有多价金属阳离子的化合物。
[0236]
(混合工序)
[0237]
本工序是将吸水性树脂粉末与上述表面交联剂进行混合的工序。关于该表面交联剂的混合方法,没有特别限定,可列举出:预先制作表面交联剂溶液,对吸水性树脂粉末优选喷雾或滴加该液体,更优选喷雾混合该液体的方法。
[0238]
作为进行该混合的装置,没有特别限定,可优选列举出高速搅拌型混合机,可更优
选列举出高速搅拌型连续混合机。
[0239]
(加热处理工序)
[0240]
本工序是对从上述混合工序中排出的混合物施加热,使吸水性树脂粉末的表面上发生交联反应的工序。
[0241]
作为进行该交联反应的装置,没有特别限定,可优选列举出桨式干燥器。该交联反应中的反应温度根据所使用的表面交联剂的种类来适当设定,优选为50~300℃、更优选为100~200℃。
[0242]
(冷却工序)
[0243]
本工序是在上述加热处理工序后根据需要而设置的任选工序。
[0244]
作为进行该冷却的装置,没有特别限定,优选为规格与加热处理工序中使用的装置相同的装置,更优选为桨式干燥器。这是因为:通过将热介质变更为制冷剂,从而能够用作冷却装置。需要说明的是,通过上述加热处理工序而得到的吸水性树脂颗粒在该冷却工序中根据需要优选强制冷却至40~80℃、更优选强制冷却至50~70℃。
[0245]
(3-7)添加剂添加工序
[0246]
本工序是向通过上述表面交联工序而得到的吸水性树脂颗粒中添加水溶性的含有多价金属阳离子的化合物、多价金属盐、阳离子性聚合物、螯合剂、无机还原剂、羟基羧酸化合物、水不溶性无机颗粒、表面活性剂、非高分子水溶性化合物等添加剂的工序。如上所述那样,也可以将该添加剂和上述表面交联剂(水溶液)同时与吸水性树脂粉末进行混合。
[0247]
(多价金属盐和/或阳离子性聚合物)
[0248]
从提高所得吸水性树脂的吸水速度、通液性、吸湿流动性等的观点出发,可以添加多价金属盐和/或阳离子性聚合物。
[0249]
作为上述多价金属盐和/或阳离子性聚合物,具体而言,在本发明中应用国际公开第2011/040530号的“〔7〕多价金属盐和/或阳离子性聚合物”所公开的化合物及其用量。
[0250]
尤其是如上所述那样,从容易获得满足式(1)的颗粒状吸水剂的方面出发,向通过上述表面交联工序而得到的吸水性树脂颗粒中添加水溶性的含有多价金属阳离子的化合物的形态是优选形态。
[0251]
(螯合剂)
[0252]
从所得吸水性树脂的色调(抗着色)、抗劣化等观点出发,可以添加螯合剂。
[0253]
作为上述螯合剂,具体而言,在本发明中应用国际公开第2011/040530号的“〔2〕螯合剂”所公开的化合物及其用量。
[0254]
(无机还原剂)
[0255]
从所得吸水性树脂的色调(抗着色)、抗劣化、降低残留单体等观点出发,可以添加无机还原剂。
[0256]
作为上述无机还原剂,具体而言,在本发明中应用国际公开第2011/040530号的“〔3〕无机还原剂”所公开的化合物及其用量。
[0257]
(α-羟基羧酸化合物)
[0258]
从所得吸水性树脂的色调(抗着色)等观点出发,可以添加α-羟基羧酸。需要说明的是,“α-羟基羧酸化合物”是指在分子内具有羟基的羧酸或其盐,是在α位具有羟基的羟基羧酸。
[0259]
作为上述α-羟基羧酸化合物,具体而言,在本发明中应用国际公开第2011/040530号的“〔6〕α-羟基羧酸化合物”所公开的化合物及其用量。
[0260]
(水不溶性无机颗粒)
[0261]
从改善吸水性树脂的流动性等观点出发,可以添加水不溶性无机颗粒。具体而言,可列举出在上述(2-9)一栏中记载的水不溶性无机颗粒。如上所述那样,从容易获得满足式(1)的颗粒状吸水剂的方面出发,向通过上述表面交联工序而得到的吸水性树脂颗粒中添加水不溶性无机颗粒的形态是优选形态。
[0262]
(表面活性剂)
[0263]
从提高所得吸水性树脂的物性(例如吸水速度)等观点出发,可以添加表面活性剂。
[0264]
作为上述表面活性剂,具体而言,可列举出国际公开第97/017397号、美国专利第6107358号中公开的表面活性剂,即,非离子性表面活性剂、阴离子性表面活性剂、阳离子性表面活性剂、两性表面活性剂等。
[0265]
(非高分子水溶性化合物)
[0266]
从降低吸水性树脂的粉尘等观点出发,可以添加非高分子水溶性化合物。在本发明中应用国际公开第2014/034667号的“非高分子水溶性化合物”所公开的化合物及其用量。
[0267]
本发明中,为了对吸水性树脂附加各种功能,也可以添加除上述添加剂之外的添加剂。作为该添加剂,具体而言,可列举出具有磷原子的化合物、氧化剂、有机还原剂、金属皂等有机粉末、消臭剂、抗菌剂、纸浆、热塑性纤维等。
[0268]
该添加剂的用量(添加量)可根据其用途来适当决定,因此没有特别限定,相对于吸水性树脂100重量份,优选为3重量份以下、更优选为1重量份以下。另外,该添加剂也可以在与上述工序不同的工序中添加。
[0269]
(3-8)其它工序
[0270]
本发明中,除了上述工序之外,可以根据需要而设置造粒工序、整粒工序、微粉去除工序、微粉的再利用工序等。另外,也可以进一步包括运输工序、贮藏工序、捆包工序、保管工序等中的1种或2种以上的工序。需要说明的是,“整粒工序”包括:在表面交联工序中及之后的微粉去除工序;在吸水性树脂发生聚集而超过期望大小的情况下,进行分级、粉碎的工序。另外,“微粉的再利用工序”中,除了如本发明那样地直接添加微粉的形态之外,还包括:制成大的含水凝胶并在吸水性树脂的制造工序中的任意工序中进行添加的工序。
[0271]
〔4〕颗粒状吸水剂的用途
[0272]
本发明的颗粒状吸水剂可用于以吸水为目的的用途,广泛用作吸收体。另外,用作包含该吸收体的吸收物品。尤其是,本发明的粒状吸水剂在加压下的倒流得以降低,因此,在吸收物品之中,可适合地用作人所使用的用于吸收尿、血液等体液的卫生物品。
[0273]
即,本发明的一个适合实施方式是包含上述形态的颗粒状吸水剂的吸收体。
[0274]
另外,本发明的另一个适合实施方式是包含上述形态的吸收体的卫生物品。
[0275]
作为吸收体,可列举出以颗粒状吸水剂和纤维基材(例如亲水性纤维)作为主成分并成型得到的吸收材料。进一步优选上述吸收体中的颗粒状吸水剂的含量(芯浓度)相对于颗粒状吸水剂与亲水性纤维的总重量为20~100重量%、更优选为25~90重量%、特别优选
为30~80重量%、最优选为40~80重量%。上述吸收体中的芯浓度越高,则在制造吸收体、吸收物品等时越会受到颗粒状吸水剂的吸水性能的影响。这种吸收体例如是对亲水性纤维等纤维基材与颗粒状吸水剂进行共混或夹心而成形的。作为所使用的纤维基材,可列举出:例如,经粉碎的木材纸浆等亲水性纤维;棉籽绒、交联纤维素纤维、人造丝、棉、羊毛、乙酸酯、维尼纶等。这些纤维基材优选为进行气流成网而成的物质。
[0276]
另外,作为吸收体,可以为在两个片(例如非织造布)之间固定吸水性树脂而得到的(无浆的)吸水性片。
[0277]
另外,上述吸收物品是指具备上述吸收体、具有透液性的表面片和具有不透液性的背面片而成的吸收物品。关于上述吸收性物品,制造吸收体(吸收芯),将该吸收芯用具有透液性的表面片和具有不透液性的背面片夹持。其后,根据需要,通过装备弹性构件、扩散层、粘合带等,从而得到成人用纸尿布、生理用卫生巾等吸收物品。需要说明的是,此时,上述吸收芯被压缩成形至例如密度为0.06~0.50[g/cm3]、基重为0.01~0.20[g/cm2]的范围。
[0278]
实施例
[0279]
使用以下的实施例和比较例,更详细地说明本发明。其中,本发明的技术范围不仅仅限定于以下的实施例。另外,在下述实施例中,只要没有特别记载,则在室温(20~25℃)/相对湿度为45~55%rh的条件下进行操作。
[0280]
需要说明的是,实施例、比较例和参照例中使用的电气设备(还包括颗粒状吸水剂的物性测定)只要没有特别注释,就使用200v或100v的电源。
[0281]
(a)吸湿结块率(b.r.;blocking ratio)的测定方法
[0282]
将颗粒状吸水剂或吸水性树脂2g均匀地散布至直径52mm的铝杯后,在温度为25℃、相对湿度为90
±
5%rh下的恒温恒湿机(platinous luciferpl-2g;tabai espec公司制)中静置1小时。在经过1小时后,将上述铝杯中盛装的颗粒状吸水剂或吸水性树脂轻轻地转移至网眼为2000μm(jis8.6目)的jis标准筛(the iida testing sieve:内径为80mm)上,使用ro-tap型振筛机(株式会社饭田制作所制、es-65型振筛机;转速为230rpm、冲击数为130rpm),在室温(20~25℃)、相对湿度为50%rh的条件下分级5秒钟。测定残留在上述jis标准筛上的颗粒状吸水剂或吸水性树脂的重量(w1[g])以及穿过了该jis标准筛的颗粒状吸水剂或吸水性树脂的重量(w2[g]),按照下式,计算吸湿流动性(吸湿结块率)。需要说明的是,结块率的值越低,则吸湿流动性越优异。
[0283]
吸湿流动性(b.r.)[重量%]={w1/(w1 w2)}
×
100。
[0284]
(b)粉尘量的测定方法
[0285]
按照国际公开第2006/098271号的[281]~[282]的记载来实施。即,根据利用下述条件在规定时间内被玻璃纤维滤纸吸引捕获的灰尘的增重,测定颗粒状吸水剂的粉尘量。作为测定装置,利用德国heubach engineering gmbh制的海博灰尘测试仪(heubach dustmeter),在测定模式为typeii的条件下来实施。测定时的气氛温度为23℃(
±
2℃)、相对湿度为20~40%rh,在常压下进行。测定方法如下那样地进行。
[0286]
(1)向转筒中装入作为测定样品的颗粒状吸水剂100.00g。
[0287]
(2)将保留粒径为0.5μm(jis p3801)且直径为50mm的玻璃纤维滤纸(例如将advantec公司制、glass fiber、gc-90或其相应制品加工成直径50mm)的重量测定至0.00001g单位([da]g)。
[0288]
(3)对转筒安装大型颗粒分离机,安装配有玻璃纤维滤纸的过滤器盒。
[0289]
(4)如下述那样地设定灰尘测试仪中的控制部的测定条件,并进行测定。筒转速:30rpm、抽吸风量:4l/min、time(测定时间):30分钟。
[0290]
(5)在规定时间后,将玻璃纤维滤纸的重量测定至0.00001g单位([db])。
[0291]
使用前述da和前述db,粉尘量按照下述式(8)来计算。
[0292]
粉尘量[mg/kg]=([db]-[da])/100
×
1000000式(8)。
[0293]
(c)表面张力的测定方法
[0294]
向充分清洗的100ml烧杯中投入调整至20℃的生理盐水50ml,首先,使用表面张力计(kruss公司制的k11自动表面张力计),测定生理盐水的表面张力。在该测定中,表面张力的值必须为71~75[mn/m]的范围。
[0295]
接着,向包含调整至20℃且测定表面张力后的生理盐水的烧杯中投入充分清洗的25mm长的氟树脂制转子以及颗粒状吸水剂或吸水性树脂0.5g,在500rpm的条件下搅拌4分钟。在4分钟后,停止搅拌,在含水的颗粒状吸水剂或吸水性树脂发生沉降后,再次进行相同的操作来测定上清液的表面张力。需要说明的是,本发明中,采取使用白金板的板法,板在各测定前充分用去离子水进行清洗,且利用气体燃烧器进行加热清洗来使用。
[0296]
(d)着色评价(黄色度/yi值)
[0297]
颗粒状吸水剂或吸水性树脂的着色评价使用日本电色工业公司制的分光式色差计sz-σ80color measuring system。在设定条件(反射测定/附带的粉末/糊剂试样台(内径为30mm、高度为12mm/作为标准的粉末/糊剂用标准圆白板no.2/30φ投光管))下,将5g颗粒状吸水剂或吸水性树脂填充至所配备的试样台中(填充所配备的试样台的6成左右),在室温(20~25℃)、湿度为50rh%的条件下,利用上述分光式色差计来测定表面颜色(yi值(yellow index))。另外,利用相同装置的相同测定方法,也可以同时测定其它尺度的物体颜色(l,a,b)或wb(亨特色度)。l/wb越大、a/b越小,则表示着色越低而实质上越接近白色。
[0298]
接着,向上述糊剂试样台中填充5g颗粒状吸水剂或吸水性树脂,将在温度调整至70
±
1℃、相对湿度调整至65
±
1%rh的气氛的恒温恒湿机(espec公司制、品名:小型环境试验器、型号:sh-641)中填充有颗粒状吸水剂或吸水性树脂的糊剂试样台暴露14天。在曝露后,利用上述分光式色差计来测定表面颜色(yi值(yellow index))。yi值优选为35以下、更优选为32以下、进一步优选为29以下、特别优选为26以下。
[0299]
(e)油漆搅拌器测试
[0300]
向直径6cm、高度11cm的玻璃制容器中投入吸水性树脂30g,设置于油漆搅拌器(no.488试验用分散机、株式会社东洋精机制作所制)中。接着,使油漆搅拌器以800(cycle/min)振荡规定时间后,使其停止。
[0301]
[制造例1]
[0302]
向容量2升的聚丙烯制容器中投入丙烯酸351.7g、作为内部交联剂的聚乙二醇二丙烯酸酯(分子量为523)0.860g(相对于含有羧基的不饱和单体为0.034摩尔%)、1.0重量%的二乙烯三胺五乙酸三钠(dtpa
·
3na)水溶液2.15g、48.5重量%的氢氧化钠水溶液149.0g和去离子水(离子交换水)336.2g并使其混合,制作单体水溶液(a’)。
[0303]
接着,将上述单体水溶液(a’)边搅拌边冷却。在液体温度成为40.0℃的时刻,添加调温至40℃的48.5重量%的氢氧化钠水溶液144.8g,并进行混合,由此制作单体水溶液
(a)。此时,该单体水溶液(a)的温度因刚刚制作后的第二阶段的中和热而上升至78.2℃。在刚刚开始混合48.5重量%的氢氧化钠水溶液后就观察到析出物,但逐渐溶解而形成透明的均匀溶液。
[0304]
接着,向搅拌状态的上述单体水溶液(a)中添加4.0重量%的过硫酸钠水溶液15.49g后,立即在大气开放体系中注入至不锈钢制托盘型容器(底面为340
×
340mm、高度为25mm、内表面:特氟隆(注册商标)涂层)内。需要说明的是,从开始第二阶段的中和起至向托盘型容器中注入单体水溶液(a)为止的时间设为55秒钟,使用加热板(neo hotplate hi-1000/株式会社井内盛荣堂社),将该托盘型容器加热至表面温度达到40℃为止。
[0305]
上述单体水溶液(a)在被注入至托盘型容器中起经过60秒后,开始聚合反应。该聚合反应中,在边产生水蒸气边朝着四面八方膨胀发泡而进行后,收缩至比托盘型容器略大的尺寸。自开始聚合反应起经过3分钟后,取出含水凝胶状交联聚合物(以下称为“含水凝胶”)(1)。需要说明的是,这些一系列的操作在大气开放体系中进行。
[0306]
将上述聚合反应中得到的含水凝胶(1)切割成短条状,供给至螺杆挤出机并进行凝胶粉碎,得到颗粒状含水凝胶(1)。需要说明的是,在螺杆挤出机中,在前端部具备直径100mm、孔径11.0mm、孔数40个、开孔率62.5%、厚度10mm的多孔板,螺杆轴的外径为86mm。
[0307]
通过在将上述螺杆挤出机的螺杆轴的转速设为130rpm的状态下,分别从不同的供给口同时供给上述短条状的含水凝胶(1)和水蒸气来进行上述凝胶的粉碎。需要说明的是,该含水凝胶(1)的供给量为每分钟4640g、水蒸气的供给量为每分钟83g。
[0308]
将该颗粒状含水凝胶(1)铺展在50目的金属网上,以190℃进行30分钟的热风干燥,将干燥物用辊磨机(wml型辊粉碎机/有限会社井口技研社)进行粉碎,进而,用具有网眼为850μm、600μm、500μm、300μm、150μm的jis标准筛进行筛分后,进行调配,由此得到重均粒径(d50)为305μm、粒度分布的对数标准偏差(σζ)为0.35的不规则破碎状的前体吸水性树脂(a)。前体吸水性树脂(a)的离心分离机保持容量(crc)为48.4(g/g)。
[0309]
[制造例2]
[0310]
向容量2升的聚丙烯制容器中投入丙烯酸335.3g、作为内部交联剂的聚乙二醇二丙烯酸酯(分子量为523)0.720g(相对于含有羧基的不饱和单体为0.030摩尔%)、1.0重量%的二乙烯三胺五乙酸三钠(dtpa
·
3na)水溶液2.05g、48.5重量%的氢氧化钠水溶液142.1g和去离子水(离子交换水)367.2g并使其混合,制作单体水溶液(b’)。
[0311]
接着,将上述单体水溶液(b’)边搅拌边冷却。在液体温度成为42.0℃的时刻,添加调温至40℃的48.5重量%的氢氧化钠水溶液138.1g,并进行混合,由此制作单体水溶液(b)。此时,该单体水溶液(b)的温度因刚刚制作后的第二阶段的中和热而上升至77.8℃。在刚刚开始混合48.5重量%的氢氧化钠水溶液后就观察到析出物,但逐渐溶解而形成透明的均匀溶液。
[0312]
接着,向搅拌状态的上述单体水溶液(b)中添加4.0重量%的过硫酸钠水溶液14.77g后,立即在大气开放体系中注入至不锈钢制托盘型容器(底面为340
×
340mm、高度为25mm、内表面:特氟隆(注册商标)涂层)内。需要说明的是,从开始第二阶段的中和起至向托盘型容器中注入单体水溶液(b)为止的时间设为55秒钟,使用加热板(neo hotplate hi-1000/株式会社井内盛荣堂社),将该托盘型容器加热至表面温度达到40℃为止。
[0313]
上述单体水溶液(b)在被注入至托盘型容器中起经过60秒后,开始聚合反应。该聚
合反应中,在边产生水蒸气边朝着四面八方膨胀发泡而进行后,收缩至比托盘型容器略大的尺寸。自开始聚合反应起经过3分钟后,取出含水凝胶状交联聚合物(以下称为“含水凝胶”)(2)。需要说明的是,这些一系列的操作在大气开放体系中进行。
[0314]
将上述聚合反应中得到的含水凝胶(2)切割成短条状,供给至螺杆挤出机并进行凝胶粉碎,得到颗粒状含水凝胶(2)。需要说明的是,在螺杆挤出机中,在前端部具备直径100mm、孔径9.5mm、孔数40个、开孔率62.5%、厚度10mm的多孔板,螺杆轴的外径为86mm。
[0315]
通过在将上述螺杆挤出机的螺杆轴的转速设为130rpm的状态下,分别从不同的供给口同时供给上述短条状的含水凝胶(2)和水蒸气来进行上述凝胶的粉碎。需要说明的是,该含水凝胶(2)的供给量为每分钟4640g、水蒸气的供给量为每分钟83g。
[0316]
将该细分化的含水凝胶(2)铺展在50目的金属网上,以190℃进行30分钟的热风干燥,将干燥物用辊磨机(wml型辊粉碎机/有限会社井口技研社)进行粉碎,进而,用具有网眼为850μm、600μm、500μm、300μm、150μm的jis标准筛进行筛分后,进行调配,由此得到重均粒径(d50)为298μm、粒度分布的对数标准偏差(σζ)为0.35的不规则破碎状的前体吸水性树脂(b)。前体吸水性树脂(b)的离心分离机保持容量(crc)为50.1(g/g)。
[0317]
(实施例1-1)
[0318]
相对于前体吸水性树脂(a)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、碳酸亚乙酯0.3重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(1)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.01重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:reolosil qs-20、tokuyama公司制)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合30分钟,得到颗粒状吸水剂(1)。将颗粒状吸水剂(1)的吸收性能示于表1。另外,颗粒状吸水剂(1)的aap4.83kpa为19.4[g/g]、gpr为81[g/min]、流下速度为10.1[g/s]。
[0319]
(实施例1-2)
[0320]
相对于前体吸水性树脂(a)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、1,3-丙二醇0.26重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(2)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、乙二胺四亚甲基膦酸五钠(edtmp
·
5na)0.01重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:reolosil qs-20、tokuyama公司制)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(2)。将颗粒状吸水剂(2)的吸收性能示于表1。
[0321]
(实施例1-3)
[0322]
在制造例1中,利用具有850μm、600μm、500μm、300μm、150μm网眼的jis标准筛进行筛分后,将重均粒径(d50)调整为343μm、将粒度分布的对数标准偏差(σζ)调整为0.36,相对于由此得到的前体吸水性树脂(a)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025
重量份、1,4-丁二醇0.31重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(3)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.03重量份和聚氧乙烯(20)脱水山梨糖醇单硬脂酸酯(商品名:rheodol tw-s120v、花王公司制)0.01重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:aerosil200、日本aerosil制)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(3)。将颗粒状吸水剂(3)的吸收性能示于表1。
[0323]
(实施例1-4)
[0324]
相对于前体吸水性树脂(a)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.03重量份、丙二醇1.5重量份和去离子水3.5重量份的表面交联剂溶液,以100℃进行30分钟左右的加热处理使所得吸水性树脂(4)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.05重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:reolosil qs-20、tokuyama公司制)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(4)。将颗粒状吸水剂(4)的吸收性能示于表1。
[0325]
(实施例1-5)
[0326]
相对于前体吸水性树脂(a)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、碳酸亚乙酯0.3重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(5)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.01重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:sipernat 22s、evonik公司制)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合45分钟,得到颗粒状吸水剂(5)。将颗粒状吸水剂(5)的吸收性能示于表1。
[0327]
(实施例1-6)
[0328]
相对于前体吸水性树脂(a)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.04重量份、丙二醇2.8重量份和去离子水4.2重量份的表面交联剂溶液,以100℃进行30分钟左右的加热处理使所得吸水性树脂(6)的crc成为约35[g/g]。其后进行冷却,实施上述油漆搅拌器测试(振荡时间:15分钟),赋予与制造工艺相当的损伤后,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、乙二胺四亚甲基膦酸五钠(edtmp
·
5na)0.05重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:sipernat 22s、evonik公司制)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(6)。将颗粒状吸水剂(6)的吸收性能示于表
1。
[0329]
(实施例1-7)
[0330]
相对于前体吸水性树脂(a)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、乙二醇0.21重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(7)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水0.5重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.05重量份和聚丙二醇700(kishida chemical公司制)0.25重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:osc c132、oriental silica corporation)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(7)。颗粒状吸水剂(7)的gpr为46[g/min]、粉尘量为70[mg/kg]。另外,将颗粒状吸水剂(7)的吸收性能示于表1。
[0331]
(实施例1-8)
[0332]
相对于前体吸水性树脂(a)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、碳酸亚乙酯0.3重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(8)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.01重量份、聚氧乙烯(20)脱水山梨糖醇单硬脂酸酯(商品名:rheodol tw-s120v、花王公司制)0.01重量份和聚乙二醇400(商品名:xg-40a、日本触媒公司制)0.2重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:osc c132、oriental silica corporation)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(8)。将颗粒状吸水剂(8)的吸收性能示于表1。
[0333]
(实施例1-9)
[0334]
在制造例1中,利用具有850μm、600μm、500μm、300μm、150μm网眼的jis标准筛进行筛分后,将重均粒径(d50)调整为379μm、将粒度分布的对数标准偏差(σζ)调整为0.38,相对于由此得到的前体吸水性树脂(a)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.03重量份、丙二醇1.2重量份和去离子水2.8重量份的表面交联剂溶液,以90℃进行30分钟左右的加热处理使所得吸水性树脂(9)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.01重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合氢氧化铝(富士胶片和光纯药公司制)0.4重量份。在混合中,将吸水性树脂30g与氢氧化铝一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(9)。颗粒状吸水剂(9)的aap4.83kpa为22.1[g/g]、表面张力为72.4mn/m。另外,将颗粒状吸水剂(9)的吸收性能示于表1。
[0335]
(实施例1-10)
[0336]
相对于前体吸水性树脂(a)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.04重量份、丙二醇2.45重量份、去离子水3.55重量份和硫酸铝14~18水合物0.75重量份的表
面交联剂溶液,以100℃进行30分钟左右的加热处理使所得吸水性树脂(10)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.03重量份和聚乙二醇600(商品名:peg-600、三洋化成工业公司制)0.1重量份的水溶液。以60℃干燥1小时后,使其穿过网眼850μm的jis标准筛。进而,将吸水性树脂30g投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(10)。颗粒状吸水剂(10)的aap4.83kpa为18.5[g/g]、吸湿结块率为0[%]、粉尘量为260[mg/kg]。另外,将颗粒状吸水剂(10)的吸收性能示于表1。
[0337]
(实施例1-11)
[0338]
相对于前体吸水性树脂(b)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、1,3-丙二醇0.26重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(11)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.03重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:reolosil qs-20、tokuyama公司制)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合30分钟,得到颗粒状吸水剂(11)。将颗粒状吸水剂(11)的吸收性能示于表1。
[0339]
(实施例1-12)
[0340]
相对于前体吸水性树脂(b)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、碳酸亚乙酯0.3重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(12)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、乙二胺四亚甲基膦酸五钠(edtmp
·
5na)0.1重量份、聚氧乙烯(20)脱水山梨糖醇单硬脂酸酯(商品名:rheodol tw-s120v、花王公司制)0.01重量份和聚乙二醇1000(商品名:peg-1000、三洋化成工业公司制)0.2重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:reolosil qs-20、tokuyama公司制)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(12)。颗粒状吸水剂(12)的gpr为71[g/min]、粉尘量为150[mg/kg]。另外,将颗粒状吸水剂(12)的吸收性能示于表1。
[0341]
(实施例1-13)
[0342]
相对于前体吸水性树脂(b)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、1,4-丁二醇0.31重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(13)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.03重量份和聚乙二醇400(商品名:xg-40a、日本触媒公司制)0.1重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:aerosil200、日本aerosil公司)0.3重量份。在混合中,将吸水性树脂30g与二氧
化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(13)。将颗粒状吸水剂(13)的吸收性能示于表1。
[0343]
(实施例1-14)
[0344]
相对于前体吸水性树脂(b)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.03重量份、丙二醇1.5重量份和去离子水3.5重量份的表面交联剂溶液,以100℃进行30分钟左右的加热处理使所得吸水性树脂(14)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、乙二胺四亚甲基膦酸五钠(edtmp
·
5na)0.05重量份和聚乙二醇1000(商品名:peg-1000、三洋化成工业公司制)0.05重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:reolosil qs-20、tokuyama公司制)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合30分钟,得到颗粒状吸水剂(14)。将颗粒状吸水剂(14)的吸收性能示于表1。
[0345]
(实施例1-15)
[0346]
相对于前体吸水性树脂(b)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、碳酸亚乙酯0.3重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(15)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.03重量份和聚氧乙烯(20)脱水山梨糖醇单硬脂酸酯(商品名:rheodol tw-s120v、花王公司制)0.01重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:sipernat 22s、evonik公司制)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),混合60分钟,得到颗粒状吸水剂(15)。将颗粒状吸水剂(15)的吸收性能示于表1。
[0347]
(实施例1-16)
[0348]
相对于前体吸水性树脂(b)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.04重量份、丙二醇2.8重量份和去离子水4.2重量份的表面交联剂溶液,以100℃进行30分钟左右的加热处理使所得吸水性树脂(16)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、乙二胺四亚甲基膦酸五钠(edtmp
·
5na)0.01重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:sipernat 22s、evonik公司制)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(16)。将颗粒状吸水剂(16)的吸收性能示于表1。
[0349]
(实施例1-17)
[0350]
相对于前体吸水性树脂(b)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、乙二醇0.21重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(17)的crc成为约35[g/g]。其后
进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.05重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:osc c132、oriental silica corporation)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),混合60分钟,得到颗粒状吸水剂(17)。将颗粒状吸水剂(17)的吸收性能示于表1。
[0351]
(实施例1-18)
[0352]
相对于前体吸水性树脂(b)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、碳酸亚乙酯0.3重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(18)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.01重量份、聚氧乙烯(20)脱水山梨糖醇单硬脂酸酯(商品名:rheodol tw-s120v、花王公司制)0.001重量份和聚乙二醇600(商品名:peg-600、三洋化成工业公司制)0.2重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:osc c132、oriental silica corporation)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(18)。将颗粒状吸水剂(18)的吸收性能示于表1。
[0353]
(实施例1-19)
[0354]
相对于前体吸水性树脂(a)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.04重量份、丙二醇4.0重量份、去离子水5.8重量份和硫酸铝14~18水合物(富士胶片和光纯药公司制)0.75重量份的表面交联剂溶液,以100℃进行30分钟左右的加热处理使所得吸水性树脂(19)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、乙二胺四亚甲基膦酸五钠(edtmp
·
5na)0.05重量份和聚丙二醇700(kishida chemical公司制)0.05重量份的水溶液。以60℃干燥1小时后,使其穿过网眼850μm的jis标准筛。进而,将吸水性树脂30g投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(19)。颗粒状吸水剂(19)的aap4.83kpa为19.5[g/g]、gpr为114[g/min]、吸湿结块率为0[%]。另外,将颗粒状吸水剂(19)的吸收性能示于表1。
[0355]
(实施例1-20)
[0356]
在制造例1中,利用具有850μm、600μm、500μm、300μm、150μm网眼的jis标准筛进行筛分后,将重均粒径(d50)调整为379μm、将粒度分布的对数标准偏差(σζ)调整为0.38,相对于由此得到的前体吸水性树脂(a)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.03重量份、丙二醇1.2重量份和去离子水2.8重量份的表面交联剂溶液,以90℃进行30分钟左右的加热处理使所得吸水性树脂(20)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1.5重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.01重量份、硫酸铝14~18水合物(富士胶片和光纯药公司制)0.75重量份和丙二醇0.75重量份的水溶液。以60℃干燥1小时后,使其穿过网眼850μm的jis标准筛。进而,将吸水性树脂30g投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公
司制),混合60分钟,得到颗粒状吸水剂(20)。颗粒状吸水剂(20)的aap4.83kpa为22.9[g/g]。另外,将颗粒状吸水剂(20)的吸收性能示于表1。
[0357]
(实施例1-21)
[0358]
将前体吸水性树脂(a)50重量份添加至填充有去离子水的99l塑料桶中,静置10分钟,在确认凝胶沉降后,在塑料桶上覆盖网眼150μm的筛网片,并固定于塑料桶。在筛网片上固定软管,通过软管向塑料桶内通入3小时的去离子水。在通水后,使用网眼150μm的jis标准筛对塑料桶内的液体进行控液,将所得凝胶铺展在50目的金属网上,以60℃风干24小时后,以60℃进行减压干燥,直至含水率达到7%为止。
[0359]
将干燥物用辊磨机(wml型辊粉碎机/有限会社井口技研社)进行粉碎,进而,用具有网眼为850μm、600μm、500μm、300μm、150μm的jis标准筛进行筛分后,进行调配,由此得到重均粒径(d50)为314μm、粒度分布的对数标准偏差(σζ)为0.35的不规则破碎状的前体吸水性树脂(c)。前体吸水性树脂(c)的离心分离机保持容量(crc)为49.8(g/g)。
[0360]
相对于前体吸水性树脂(c)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、碳酸亚乙酯0.3重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(21)的crc成为约35[g/g],得到颗粒状吸水剂(21)。将颗粒状吸水剂(21)的吸收性能示于表1。
[0361]
(比较例1-1)
[0362]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例1-1相同的方法,得到比较颗粒状吸水剂(1)。比较颗粒状吸水剂(1)的aap4.83kpa为18.0[g/g]、gpr为112[g/min]、流下速度为10.1[g/s]。另外,将比较颗粒状吸水剂(1)的吸收性能示于表1。
[0363]
(比较例1-2)
[0364]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例1-2相同的方法,得到比较颗粒状吸水剂(2)。将比较颗粒状吸水剂(2)的吸收性能示于表1。
[0365]
(比较例1-3)
[0366]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例1-4相同的方法,得到比较颗粒状吸水剂(3)。将比较颗粒状吸水剂(3)的吸收性能示于表1。
[0367]
(比较例1-4)
[0368]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例1-5相同的方法,得到比较颗粒状吸水剂(4)。将比较颗粒状吸水剂(4)的吸收性能示于表1。
[0369]
(比较例1-5)
[0370]
将氢氧化铝0.4重量份设为二氧化硅(商品名:osc c132、oriental silica corporation)0.3重量份,将基于转筒摇摆混合机的混合时间设为2分钟,除此之外,利用与实施例1-9相同的方法,得到比较颗粒状吸水剂(5)。将比较颗粒状吸水剂(5)的吸收性能示于表1。另外,比较颗粒状吸水剂(5)的表面张力为72.3mn/m。
[0371]
(比较例1-6)
[0372]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例1-11相同的方法,得到比较颗粒状吸水剂(6)。将比较颗粒状吸水剂(6)的吸收性能示于表1。
[0373]
(比较例1-7)
[0374]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例1-12相同的方法,得到比较颗粒状吸水剂(7)。将比较颗粒状吸水剂(7)的吸收性能示于表1。另外,比较颗粒状吸水剂(7)的gpr为78[g/min]、粉尘量为160[mg/kg]。
[0375]
(比较例1-8)
[0376]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例1-13相同的方法,得到比较颗粒状吸水剂(8)。将比较颗粒状吸水剂(8)的吸收性能示于表1。
[0377]
(比较例1-9)
[0378]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例1-15相同的方法,得到比较颗粒状吸水剂(9)。将比较颗粒状吸水剂(9)的吸收性能示于表1。
[0379]
(比较例1-10)
[0380]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例1-16相同的方法,得到比较颗粒状吸水剂(10)。将比较颗粒状吸水剂(10)的吸收性能示于表1。
[0381]
(比较例1-11)
[0382]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例1-17相同的方法,得到比较颗粒状吸水剂(11)。将比较颗粒状吸水剂(11)的吸收性能示于表1。
[0383]
(比较例1-12)
[0384]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例1-18相同的方法,得到比较颗粒状吸水剂(12)。将比较颗粒状吸水剂(12)的吸收性能示于表1。
[0385]
(比较例1-13)
[0386]
在制造例1中,以作为内部交联剂的聚乙二醇二丙烯酸酯相对于含有羧基的不饱和单体成为0.019摩尔%的方式调整单体水溶液,利用与制造例1相同的方法,得到前体吸水性树脂(d)。前体吸水性树脂(d)的离心分离机保持容量(crc)为52.7(g/g)。
[0387]
在实施例1-4中,使用前体吸水性树脂(d),将表面交联剂溶液的乙二醇变更为0.25重量份,将向吸水性树脂中添加的去离子水的量变更为10重量份,将二乙烯三胺五乙酸三钠(dtpa
·
3na)变更为0.01重量,将二氧化硅变更为水滑石(商品名:dht-6、协和化学工业公司制),并且,将水滑石的混合量设为0.4重量份,将基于转筒摇摆混合机的混合时间设为2分钟,除此之外,利用与实施例1-4相同的方法,得到比较颗粒状吸水剂(13)。将比较颗粒状吸水剂(13)的吸收性能示于表1。
[0388]
(比较例1-14)
[0389]
在比较例1-13中,将向吸水性树脂中添加的去离子水的量变更为15重量份,变更
水滑石的种类(商品名:ht-1-nc、堺化学工业公司制),并且,将水滑石的混合量设为0.2重量份,除此之外,利用比较例1-13的方法,得到比较颗粒状吸水剂(14)。将比较颗粒状吸水剂(14)的吸收性能示于表1。
[0390]
[吸收体的吸收量的评价]
[0391]
利用以下所述的方法,制作吸收体a和b,评价吸收体的吸收量。
[0392]
模型吸收体a的制作方法
[0393]
将80mm
×
160mm的脱脂棉(例如可以使用川本产业公司制的切割棉8cm
×
16cm等)沿着面方向均匀撕裂,制作2片1.6g的脱脂棉片。接着,将吸水纸以成为200mm
×
200mm的方式进行切割,在吸水纸的中央附近配置8cm
×
16cm的框。在框内铺设调整至1.6g的脱脂棉片,用亚克力板等平整脱脂棉的上表面,从上方均匀地散布颗粒状吸水剂3.2g,进而载置1.6g的脱脂棉片,制成夹层结构。在对如此制作的吸收体整体施加10kg载荷的状态下,保持1分钟,对吸收体进行成型。其后,取下载荷和框,沿着吸收体的长度方向,将吸水纸的两端以包裹吸收体的方式进行折叠。将其放入使用heatron paper而制作的非织造布袋(10cm
×
22cm)中,将周围进行热封,制成模型吸收体a。
[0394]
模型吸收体b的制作方法
[0395]
切出100mm
×
180mm的塑料胶带(例如可以使用日东电工公司制的塑料胶带21-100tm等),使其粘合面朝上,在其中央设置80mm
×
160mm的框,向框内均匀地散布颗粒状吸水剂3.2g。将框取下,另行载置切成100mm
×
180mm尺寸的纺粘非织造布,与塑料胶带进行贴合。将其放入使用heatron paper而制作的非织造布袋(10cm
×
22cm)中,将周围进行热封,制成模型吸收体b。
[0396]
吸收量测定方法
[0397]
向深型托盘中投入0.9重量%氯化钠水溶液,使其液体深度达到5cm以上,以托盘中的液体温度成为37℃的方式进行调温。在该托盘中浸渍模型吸收体,在不施加载荷的状态下使其溶胀60分钟。在浸渍时,模型吸收体b以塑料胶带面成为上表面的方式进行浸渍,之后的操作全部以塑料胶带面为上表面来进行。在溶胀后,从托盘中取出模型吸收体,载置于直径45cm、网眼2000μm的jis标准筛,相对于模型吸收体的存在有颗粒状吸水剂的8cm
×
16cm的面积,承载2690g的重物(21g/cm2),进行1分钟的控液。测量控液后的模型吸收体的重量,将其与预先测量的浸渍前的模型吸收体重量之差作为吸收量。将结果示于表1。
[0398]
[表1-1]
[0399][0400]
[表1-2]
[0401][0402]
针对上述实施例、比较例,相对于crc[g/g](横轴)标绘aap(2.06kpa) rcap(2.06kpa)(纵轴)而得到的图是图2。直线表示aap(2.06kpa) rcap(2.06kpa)=0.58
×
crc
55.6。
[0403]
由表1的吸收体的评价结果可知:与比较例相比,满足式(1)的颗粒状吸水剂、即实施例的颗粒状吸水剂在用于吸收体时,即便颗粒状吸水剂呈现吸收液体而发生溶胀的状态,其加压下的液体保持量(吸收量)也多1g以上。该液体保持量的差异在本领域中是显著的差异。根据该结果可知:在颗粒状吸水剂为溶胀状态时,即便颗粒状吸水剂从外部承载有压力,实施例的颗粒状吸水剂也能够显著地降低液体回流。
[0404]
[制造例3]
[0405]
向容量2升的聚丙烯制容器中投入丙烯酸351.7g、作为内部交联剂的聚乙二醇二丙烯酸酯(分子量为523)0.910g(相对于含有羧基的不饱和单体为0.036摩尔%)、1.0重量%的二乙烯三胺五乙酸三钠(dtpa
·
3na)水溶液2.15g、48.5重量%的氢氧化钠水溶液149.0g、50.0重量%的苹果酸水溶液(dl-苹果酸、50.0%水溶液、扶桑化学工业公司制、食品添加物级别)1.41g(苹果酸相对于含有羧基的不饱和单体为0.108摩尔%)和去离子水(离子交换水)336.2g并使其混合,制作单体水溶液(e’)。
[0406]
接着,将上述单体水溶液(e’)边搅拌边冷却。在液体温度成为40.0℃的时刻,添加调温至40℃的48.5重量%的氢氧化钠水溶液144.8g,并进行混合,由此制作单体水溶液(e)。此时,该单体水溶液(e)的温度因刚刚制作后的第二阶段的中和热而上升至77.9℃。在刚刚开始混合48.5重量%的氢氧化钠水溶液后就观察到析出物,但逐渐溶解而形成透明的均匀溶液。
[0407]
接着,向搅拌状态的上述单体水溶液(e)中添加4.0重量%的过硫酸钠水溶液15.49g后,立即在大气开放体系中注入至不锈钢制托盘型容器(底面为340
×
340mm、高度为25mm、内表面:特氟隆(注册商标)涂层)内。需要说明的是,从开始第二阶段的中和起至向托盘型容器中注入单体水溶液(e)为止的时间设为55秒钟,使用加热板(neo hotplate hi-1000/株式会社井内盛荣堂社),将该托盘型容器加热至表面温度达到40℃为止。
[0408]
上述单体水溶液(e)在被注入至托盘型容器中起经过60秒后,开始聚合反应。该聚合反应中,在边产生水蒸气边朝着四面八方膨胀发泡而进行后,收缩至比托盘型容器略大的尺寸。自开始聚合反应起经过3分钟后,取出含水凝胶状交联聚合物(以下称为“含水凝胶”)(3)。需要说明的是,这些一系列的操作在大气开放体系中进行。
[0409]
将上述聚合反应中得到的含水凝胶(3)切割成短条状,供给至螺杆挤出机并进行凝胶粉碎,得到颗粒状含水凝胶(3)。需要说明的是,在螺杆挤出机中,在前端部具备直径100mm、孔径11.0mm、孔数40个、开孔率62.5%、厚度10mm的多孔板,螺杆轴的外径为86mm。
[0410]
通过在将上述螺杆挤出机的螺杆轴的转速设为130rpm的状态下,分别从不同的供给口同时供给上述短条状的含水凝胶(3)和水蒸气来进行上述凝胶的粉碎。需要说明的是,该含水凝胶(3)的供给量为每分钟4640g、水蒸气的供给量为每分钟83g。
[0411]
将该颗粒状含水凝胶(3)铺展在50目的金属网上,以190℃进行30分钟的热风干燥,将干燥物用辊磨机(wml型辊粉碎机/有限会社井口技研社)进行粉碎,进而,用具有网眼为850μm、600μm、500μm、300μm、150μm的jis筛进行筛分后,进行调配,由此得到重均粒径(d50)为303μm、粒度分布的对数标准偏差(σζ)为0.36的不规则破碎状的前体吸水性树脂(e)。前体吸水性树脂(e)的离心分离机保持容量(crc)为50.2(g/g)。
[0412]
[制造例4]
[0413]
向容量2升的聚丙烯制容器中投入丙烯酸335.3g、作为内部交联剂的聚乙二醇二丙烯酸酯(分子量为523)1.344g(相对于含有羧基的不饱和单体为0.056摩尔%)、1.0重量%的二乙烯三胺五乙酸三钠(dtpa
·
3na)水溶液2.05g、48.5重量%的氢氧化钠水溶液142.1g、50.0重量%的苹果酸水溶液(dl-苹果酸、50.0%水溶液、扶桑化学工业公司制、食品添加物级别)6.706g(苹果酸相对于含有羧基的不饱和单体为0.537摩尔%)和去离子水(离子交换水)367.2g并使其混合,制作单体水溶液(f’)。
[0414]
接着,将上述单体水溶液(f’)边搅拌边冷却。在液体温度成为42.0℃的时刻,添加调温至40℃的48.5重量%的氢氧化钠水溶液138.1g,并进行混合,由此制作单体水溶液(f)。此时,该单体水溶液(f)的温度因刚刚制作后的第二阶段的中和热而上升至78.1℃。在刚刚开始混合48.5重量%的氢氧化钠水溶液后就观察到析出物,但逐渐溶解而形成透明的均匀溶液。
[0415]
接着,向搅拌状态的上述单体水溶液(f)中添加4.0重量%的过硫酸钠水溶液14.77g后,立即在大气开放体系中注入至不锈钢制托盘型容器(底面为340
×
340mm、高度为25mm、内表面:特氟隆(注册商标)涂层)。需要说明的是,从开始第二阶段的中和起至向托盘型容器中注入单体水溶液(f)为止的时间设为55秒钟,使用加热板(neo hotplate hi-1000/株式会社井内盛荣堂社),将该托盘型容器加热至表面温度达到40℃为止。
[0416]
上述单体水溶液(f)在被注入至托盘型容器中起经过60秒后,开始聚合反应。该聚合反应中,在边产生水蒸气边朝着四面八方膨胀发泡而进行后,收缩至比托盘型容器略大的尺寸。自开始聚合反应起经过3分钟后,取出含水凝胶状交联聚合物(以下称为“含水凝胶”)(4)。需要说明的是,这些一系列的操作在大气开放体系中进行。
[0417]
将上述聚合反应中得到的含水凝胶(4)切割成短条状,供给至螺杆挤出机并进行凝胶粉碎,得到颗粒状含水凝胶(4)。需要说明的是,在螺杆挤出机中,在前端部具备直径100mm、孔径9.5mm、孔数40个、开孔率62.5%、厚度10mm的多孔板,螺杆轴的外径为86mm。
[0418]
通过在将上述螺杆挤出机的螺杆轴的转速设为130rpm的状态下,分别从不同的供给口同时供给上述短条状的含水凝胶(4)和水蒸气来进行上述凝胶的粉碎。需要说明的是,该含水凝胶(4)的供给量为每分钟4640g、水蒸气的供给量为每分钟83g。
[0419]
将该细分化的含水凝胶(4)铺展在50目的金属网上,以190℃进行30分钟的热风干燥,将干燥物用辊磨机(wml型辊粉碎机/有限会社井口技研社)进行粉碎,进而,用具有网眼为850μm、600μm、500μm、300μm、150μm的jis筛进行筛分后,进行调配,由此得到重均粒径(d50)为303μm、粒度分布的对数标准偏差(σζ)为0.35的不规则破碎状的前体吸水性树脂(f)。前体吸水性树脂(f)的离心分离机保持容量(crc)为49.6(g/g)。
[0420]
(实施例2-1)
[0421]
相对于前体吸水性树脂(e)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、碳酸亚乙酯0.3重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(22)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、乙二胺四亚甲基膦酸五钠(edtmp
·
5na)0.05重量份和亚硫酸钠0.15重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:reolosil qs-20、tokuyama公司制)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量
225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合30分钟,得到颗粒状吸水剂(22)。颗粒状吸水剂(22)的gpr为84[g/min]、表面张力为72.3mn/m、着色评价后的yi值为24。将颗粒状吸水剂(22)的吸收性能示于表2。
[0422]
(实施例2-2)
[0423]
相对于前体吸水性树脂(e)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、乙二醇0.21重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(23)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.03重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:osc c132、oriental silica corporation)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(23)。将颗粒状吸水剂(23)的吸收性能示于表2。
[0424]
(实施例2-3)
[0425]
相对于前体吸水性树脂(e)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、丙二醇1.2重量份和去离子水2.8重量份的表面交联剂溶液,以100℃进行30分钟左右的加热处理使所得吸水性树脂(24)的crc成为约35[g/g]。其后进行冷却,实施上述油漆搅拌器测试(振荡时间:10分钟),赋予与制造工艺相当的损伤后,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.01重量份和聚乙二醇600(商品名:peg-600、三洋化成工业公司制)0.2重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:sipernat 22s、evonik公司制)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(24)。将颗粒状吸水剂(24)的吸收性能示于表2。
[0426]
(实施例2-4)
[0427]
相对于前体吸水性树脂(e)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.04重量份、丙二醇3.5重量份、去离子水5.0重量份和硫酸铝14~18水合物0.75重量份的表面交联剂溶液,以100℃进行30分钟左右的加热处理使所得吸水性树脂(25)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.01重量份和聚丙二醇700(kishida chemical公司制)0.05重量份的水溶液。以60℃干燥1小时后,使其穿过网眼850μm的jis标准筛。进而,将吸水性树脂30g投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(25)。颗粒状吸水剂(25)的aap4.83kpa为19.1[g/g]、gpr为79[g/min]、吸湿结块率为0[%]。将颗粒状吸水剂(25)的吸收性能示于表2。
[0428]
(实施例2-5)
[0429]
相对于前体吸水性树脂(f)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.025重量份、碳酸亚乙酯0.3重量份、丙二醇0.5重量份和去离子水2.0重量份的表面交联剂溶液,以190℃进行30分钟左右的加热处理使所得吸水性树脂(26)的crc成为约35[g/g]。
其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、乙二胺四亚甲基膦酸五钠(edtmp
·
5na)0.1重量份、聚氧乙烯(20)脱水山梨糖醇单硬脂酸酯(商品名:rheodol tw-s120v、花王公司制)0.01重量份和聚乙二醇1000(商品名:peg-1000、三洋化成工业公司制)0.2重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:osc c132、oriental silica corporation)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(26)。将颗粒状吸水剂(26)的吸收性能示于表2。
[0430]
(实施例2-6)
[0431]
相对于前体吸水性树脂(f)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.04重量份、丙二醇2.8重量份和去离子水4.2重量份的表面交联剂溶液,以100℃进行30分钟左右的加热处理使所得吸水性树脂(27)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.03重量份和聚氧乙烯(20)脱水山梨糖醇单硬脂酸酯(商品名:rheodol tw-s120v、花王公司制)0.01重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合二氧化硅(商品名:sipernat 22s、evonik公司制)0.3重量份。在混合中,将吸水性树脂30g与二氧化硅一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合45分钟,得到颗粒状吸水剂(27)。颗粒状吸水剂(27)的流下速度为9.4[g/s]、着色评价后的yi值为23。将颗粒状吸水剂(27)的吸收性能示于表2。
[0432]
(实施例2-7)
[0433]
相对于前体吸水性树脂(f)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.03重量份、丙二醇1.5重量份和去离子水3.5重量份的表面交联剂溶液,以100℃进行30分钟左右的加热处理使所得吸水性树脂(28)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、乙二胺四亚甲基膦酸五钠(edtmp
·
5na)0.01重量份和亚硫酸氢钠0.1重量份的水溶液。以60℃干燥1小时后,使其穿过网眼为850μm的jis标准筛,混合氢氧化铝(富士胶片和光纯药公司制)0.4重量份。在混合中,将吸水性树脂30g与氢氧化铝一同投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(28)。将颗粒状吸水剂(28)的吸收性能示于表2。
[0434]
(实施例2-8)
[0435]
相对于前体吸水性树脂(f)100重量份,均匀地混合包含乙二醇二缩水甘油醚0.04重量份、丙二醇2.8重量份、去离子水4.2重量份和硫酸铝14~18水合物(富士胶片和光纯药公司制)0.75重量份的表面交联剂溶液,以100℃进行30分钟左右的加热处理使所得吸水性树脂(29)的crc成为约35[g/g]。其后进行冷却,相对于吸水性树脂100重量份,均匀地混合包含去离子水1重量份、二乙烯三胺五乙酸三钠(dtpa
·
3na)0.05重量份、聚乙二醇400(商品名:xg-40a、日本触媒公司制)0.3重量份和亚硫酸钠0.03重量份的水溶液。以60℃干燥1小时后,使其穿过网眼850μm的jis标准筛。进而,将吸水性树脂30g投入至容量225ml的蛋黄酱瓶中,使用转筒摇摆混合机t2f型(shinmaru enterprises公司制),以101rpm混合60分钟,得到颗粒状吸水剂(29)。颗粒状吸水剂(29)的粉尘量为50[mg/kg]、着色评价后的yi值
为25。将颗粒状吸水剂(29)的吸收性能示于表2。
[0436]
(实施例2-9)
[0437]
将基于转筒摇摆混合机的二氧化硅的混合时间设为15分钟,除此之外,利用与实施例2-1相同的方法,得到颗粒状吸水剂(30)。将颗粒状吸水剂(30)的吸水性能示于表2。
[0438]
(比较例2-1)
[0439]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例2-1相同的方法,得到比较颗粒状吸水剂(15)。比较颗粒状吸水剂(15)的gpr为100[g/min]、表面张力为72.2mn/m、着色评价后的yi值为24。将比较颗粒状吸水剂(15)的吸收性能示于表2。
[0440]
(比较例2-2)
[0441]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例2-3相同的方法,得到比较颗粒状吸水剂(16)。将比较颗粒状吸水剂(16)的吸收性能示于表2。
[0442]
(比较例2-3)
[0443]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例2-5相同的方法,得到比较颗粒状吸水剂(17)。将比较颗粒状吸水剂(17)的吸收性能示于表2。
[0444]
(比较例2-4)
[0445]
将基于转筒摇摆混合机的二氧化硅的混合时间设为2分钟,除此之外,利用与实施例2-6相同的方法,得到比较颗粒状吸水剂(18)。比较颗粒状吸水剂(18)的流下速度为9.2[g/s]、着色评价后的yi值为23。另外,将比较颗粒状吸水剂(18)的吸收性能示于表2。
[0446]
(比较例2-5)
[0447]
将从大王制纸公司制的尿不湿“goo.n pants massara sara透气男孩用l尺寸”(2020年购入)的吸收体中尽可能去除纸浆而采取的颗粒状吸水剂(不规则破碎状)25.5g填充至带有滑扣的聚乙烯袋(滑扣内侧的尺寸:70mm
×
50mm、厚度:0.04mm、容量:35ml)中。将该聚乙烯袋载置在网眼850mm、内径200mm的jis标准筛上。将该标准筛固定于振筛机as200(retsch公司制),以0.3mm的震荡幅度振荡30分钟。此时,颗粒状吸水剂所承受的加速度的计算值最大为2.2g。将振荡后的颗粒状吸水剂作为比较颗粒状吸水剂(19)。将比较颗粒状吸水剂(19)的吸水性能示于表3。
[0448]
(比较例2-6)
[0449]
将振荡时间变更为300分钟,除此之外,进行与比较例2-5相同的操作,由此得到比较颗粒状吸水剂(20)。将比较颗粒状吸水剂(20)的吸水性能示于表3。
[0450]
[表2]
[0451][0452]
[表3]
[0453][0454]
由表2的吸收体的评价结果可知:与比较例相比,满足式(1)的颗粒状吸水剂、即实施例的颗粒状吸水剂在用于吸收体时,即便颗粒状吸水剂呈现吸收液体而发生溶胀的状
态,其加压下的液体保持量(吸收量)也多2g以上。该液体保持量的差异在本领域中是显著的差异。根据该结果可知:在颗粒状吸水剂为溶胀状态时,即便颗粒状吸水剂从外部承载有压力,实施例的颗粒状吸水剂也能够显著地降低液体回流。
[0455]
需要说明的是,在任意实施例中均是:凝胶渗透速度(gel permeation rate:gpr)为20g/min以上,吸湿结块率为40重量%以下,流下速度(flow rate)为8.5g/s以上,粉尘量为400mg/kg以下,表面张力为65mn/m以上,吸水性树脂粉末为不规则破碎状。
[0456]
本技术基于2020年3月31日申请的日本专利申请日本特愿2020-064626号,其公开内容通过参照将其整体援引至本说明书中。
[0457]
产业上的可利用性
[0458]
400 装置、
[0459]
410 容器、
[0460]
411 盒、
[0461]
412 活塞、
[0462]
413a、413b 金属网、
[0463]
414 溶胀凝胶(使颗粒状吸水剂吸水而得到的物质)、
[0464]
415 孔、
[0465]
420 罐、
[0466]
421 玻璃管、
[0467]
422 带有旋塞玻璃管的l字管、
[0468]
423 液体、
[0469]
431 不锈钢制的金属网、
[0470]
432 捕集容器、
[0471]
433 上皿天平。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。