1.本发明属于药物制剂技术领域,具体涉及一种富马酸丙酚替诺福韦片及其制备方法。
背景技术:
2.乙型病毒性肝炎(乙肝)是由乙型肝炎病毒(hepatitis b virus,hbv)感染引起的,以乏力、食欲减退、恶心、呕吐、厌油、肝大及肝功能异常为主要临床症状,是一种发病率高、感染性强、严重危害人类健康的全身性感染病。乙肝已然成为严重威胁人类健康的难题。
3.富马酸丙酚替诺福韦(tenofovir alafenamide fumarate,taf),分子式:c
21h29
o5n6p
·
1/2(c4h4o4),分子量:534.50,化学名为:丙-2-基n-[(s)-({[(2r)-1-(6-氨基-9h-嘌呤-9-基)丙-2-基]-氧化}甲基)(苯氧基)磷酰基]-l-丙氨酸酯,(2e)-丁-2-烯二酸(2:1),其结构如下所示:
[0004][0005]
富马酸丙酚替诺福韦是由吉利德(gilead)公司开发上市的一种新型核苷类逆转录酶抑制剂(nrti),具有广谱抗病毒作用,属于替诺福韦的酯类前药。该药用药剂量、血浆组织浓度、起效速度、全身暴露量均优于抗乙肝药物富马酸替诺福韦二吡呋酯(viread,tdf)。并且由于taf在血药中几乎不分解,大量的taf直接输送至淋巴细胞、巨噬细胞中被分解为替诺福韦,进而磷酸化形成活性成分,因此taf相比tdf对肾脏和骨骼损害风险更低。目前该药是活性最好的抗乙肝药物。
[0006]
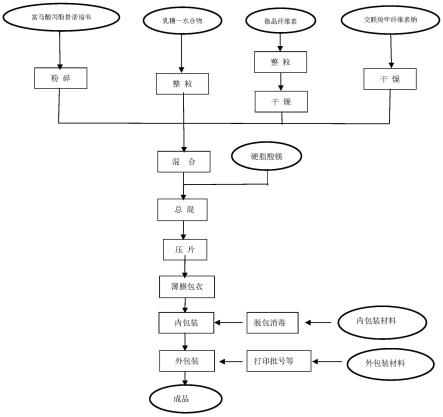
现有技术中,富马酸丙酚替诺福韦片包括活性成分、填充剂、崩解剂、粘合剂和润滑剂,经干法制粒制成。原研富马酸丙酚替诺福韦片的制备方法为:将富马酸丙酚替诺福韦(活性成分)与内加辅料(填充剂、粘合剂、崩解剂、润滑剂)混合,干法制粒,整粒后与外加辅料混合,压片,薄膜包衣,包装。原研富马酸丙酚替诺福韦片的工艺流程如附图1所示。
[0007]
原研通过干法制粒制备富马酸丙酚替诺福韦片,需要对干法制粒所得中间体颗粒进行检测控制质量,工艺过程较复杂,并且劳动强度大。而且经干法制粒机制备的薄片需要经破碎再次转化为颗粒,粉尘污染较严重,不利工人的劳动保护。此外,干法制粒设备厂房投资较高,增加了投入成本。因此,提供一种富马酸丙酚替诺福韦片,能有效克服上述问题,
成为了本领域技术人员亟待解决的问题。
技术实现要素:
[0008]
本发明的目的之一在于,提供一种富马酸丙酚替诺福韦片,解决现有技术中工艺复杂,粉尘污染重,设备厂房投入成本及检测成本高的问题。
[0009]
本发明的目的之二在于,提供该富马酸丙酚替诺福韦片的制备方法。
[0010]
为实现上述目的,本发明采用的技术方案如下:
[0011]
本发明提供了一种富马酸丙酚替诺福韦片,由包括以下重量份的组分制成:富马酸丙酚替诺福韦10-20份,乳糖一水合物35-65份,微晶纤维素12.5-45份,交联羧甲纤维素钠2-7份。
[0012]
本发明的部分实施例中,富马酸丙酚替诺福韦为14份。
[0013]
本发明的部分实施方案中,还包括有1.5~3.0份的润滑剂,优选地,所述润滑剂为硬脂酸镁。
[0014]
本发明的部分实施方案中,微晶纤维素的水分含量≤3.9wt.%,或/和交联羧甲纤维素钠的水分含量≤3.4wt.%。
[0015]
本发明的部分实施方案中,富马酸丙酚替诺福韦的粒径d90为20μm~200μm。
[0016]
本发明的部分实施方案中,还包括有薄膜衣,其中片芯与薄膜衣的质量比为100:2~4;优选地,所述薄膜衣为胃溶型薄膜衣。
[0017]
本发明的部分实施方案中,一种富马酸丙酚替诺福韦片,由包括以下重量份的组分制成:富马酸丙酚替诺福韦14份,乳糖一水合物35份,微晶纤维素45份,交联羧甲纤维素钠4.5份,硬脂酸镁1.5份;
[0018]
或,富马酸丙酚替诺福韦14份,乳糖一水合物65份,微晶纤维素15份,交联羧甲纤维素钠4.5份,硬脂酸镁1.5份;
[0019]
或,富马酸丙酚替诺福韦14份,乳糖一水合物50份,微晶纤维素30份,交联羧甲纤维素钠4.5份,硬脂酸镁1.5份;
[0020]
或,富马酸丙酚替诺福韦14份,乳糖一水合物52.5份,微晶纤维素30份,交联羧甲纤维素钠2份,硬脂酸镁1.5份;
[0021]
或,富马酸丙酚替诺福韦14份,乳糖一水合物47.5份,微晶纤维素30份,交联羧甲纤维素钠7份,硬脂酸镁1.5份;
[0022]
或,富马酸丙酚替诺福韦14份,乳糖一水合物48.5份,微晶纤维素30份,交联羧甲纤维素钠4.5份,硬脂酸镁3份。
[0023]
本发明还提供了一种富马酸丙酚替诺福韦片的制备方法,该制备方法包括以下步骤:将富马酸丙酚替诺福韦粉碎后,与乳糖一水合物、微晶纤维素、交联羧甲纤维素钠预混,加入润滑剂,总混,压片。
[0024]
本发明的部分实施方案中,分别将微晶纤维素、交联羧甲纤维素钠干燥至规定水分含量后,再与富马酸丙酚替诺福韦、乳糖一水合物混合。
[0025]
本发明的实施例中,将乳糖一水合物整粒后再预混,整粒用筛网的孔径为1.0mm。
[0026]
本发明的实施例中,将微晶纤维素整粒、干燥再预混,整粒用筛网的孔径为1.0mm,烘箱干燥,干燥温度为50℃~60℃,控制水分≤3.9wt.%。
[0027]
本发明的实施例中,将交联羧甲纤维素钠采用烘箱干燥,干燥温度为60℃~70℃,控制水分≤3.4wt.%。
[0028]
本发明的部分实施方案中,将压片制得的素片进行薄膜包衣,制得薄膜衣片。
[0029]
本发明的部分实施方案中,总混时间为5min~10min。
[0030]
本发明的部分实施方案中,压片硬度为50n/mm2~140n/mm2,优选压片硬度为80n/mm2~110n/mm2。
[0031]
与现有技术相比,本发明具有以下有益效果:
[0032]
本发明设计科学,构思巧妙,本发明的处方以富马酸丙酚替诺福韦为活性成分,并包括填充剂、粘合剂、崩解剂及润滑剂;通过粉末直压工艺制备富马酸丙酚替诺福韦的口服片剂。本发明工艺过程简单,无需制粒,所获得终产品质量较原研制剂更加稳定(杂质含量更低),批次间差异更小。
[0033]
与现有技术中的干法制粒相比,本发明减小了粉尘,减少了中间抽样检测等工序;还减少了相应设备厂房投资以及检验成本和劳动强度,节约了时间和能源,可操作性更强,连续生产更有保证,提高了临床应用的安全性,尤其适合gmp要求的生产管理,有利于该药品的商业化生产且具备广阔的市场前景。
[0034]
本发明通过对api的粒径的限定,以及辅料中占比较大的以及乳糖一水合物、微晶纤维素粒径的控制,保证了本发明片剂的批内含量均匀性且不同批次溶出始终保持一致。
[0035]
本发明通过辅料中微晶纤维素及交联羧甲纤维素钠水分含量的控制,确保了与原研品质量的一致性,并提高了产品稳定性,实验结果显示,本发明片剂的稳定性优于原研品。
附图说明
[0036]
附图1为现有技术原研药的工艺流程图;
[0037]
附图2为本发明的工艺流程图;
[0038]
附图3为本发明富马酸丙酚替诺福韦片与参比制剂溶出曲线批间差异对比图;
[0039]
附图4为本发明富马酸丙酚替诺福韦片与原研品在不同介质中的溶出曲线对比图,其中4-1为在ph1.0条件下的溶出曲线对比图;4-2为在ph4.5条件下的溶出曲线对比图;4-3为在ph6.8条件下的溶出曲线对比图;4-4为在水中的溶出曲线对比图。
具体实施方式
[0040]
以下通过具体实施方式,对本发明的上述内容做进一步的详细说明。但不应该将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术方案均属于本发明的范围。
[0041]
本发明实施例中的溶出度测定方法照《中国药典》(2015年版第四部通则0931第1法)进行检测,转速为50rpm,温度为37℃
±
0.5℃,体积为500ml。溶出液采用uv法检测,检测波长为260nm。
[0042]
本发明实施例中所述的参比制剂为美国上市原研及国内进口原研,其中参比制剂1和2为美国上市原研(商品名:)2个不同批次中的制剂,参比制剂3为国内进口原研。
[0043]
实施例1
[0044]
本实施例公开了本发明的富马酸丙酚替诺福韦片的配料处方,按mg/片计,如下表所示:
[0045]
表1配料处方列表
[0046][0047]
实施例2
[0048]
根据实施例1表1中的第1~6组配方,制备富马酸丙酚替诺福韦片,并考察制得的富马酸丙酚替诺福韦片的溶出曲线。具体的制备工艺为:
[0049]
1、前处理
[0050]
粉碎:取原料药富马酸丙酚替诺福韦经粉碎处理;控制粒径dv(90)为100μm。
[0051]
整粒:将乳糖一水合物、微晶纤维素分别用移动整粒机整粒,筛网孔径为1.0mm,备用。
[0052]
干燥:将整粒后的微晶纤维素采用烘箱干燥,干燥温度为50℃~60℃,控制水分为3.9%。将交联羧甲纤维素钠采用烘箱干燥,干燥温度为60℃~70℃,控制水分为3.4%。
[0053]
2、混合
[0054]
预混:按处方量称取前处理后的富马酸丙酚替诺福韦、乳糖一水合物、微晶纤维素及交联羧甲纤维素钠置于多向运动混合机中,混合30min。
[0055]
总混:将预混粉末与硬脂酸镁置于多向运动混合机中,混合10min。
[0056]
3、压片
[0057]
控制片重差异限度为
±
6%,硬度为80~110n/mm2。筛片除去细粉后即得富马酸丙酚替诺福韦素片。
[0058]
4、薄膜包衣
[0059]
取胃溶型薄膜包衣预混剂(opardy ii型)加适量水配制成15.0%(w/w)的包衣液,包衣增重4%。
[0060]
各组样品出曲线检测结果如下表所示:
[0061]
表2各组样品溶出曲线检测结果(ph4.5)
[0062][0063]
结论:本发明的各组样品与参比品在ph4.5介质中均快速溶出(15min溶出度均大于85%),溶出行为相似,表明各实组对应处方满足要求。
[0064]
实施例3
[0065]
本实施例考察了原料药富马酸丙酚替诺福韦的粒径范围:将原料药富马酸丙酚替诺福韦经粉碎成不同粒径后,制备成富马酸丙酚替诺福韦片,以溶出度为指标,考察其与参比制剂的一致性。配料处方同实施例1表1中第3组的处方,原料药粒径如表3,其余的具体的制备工艺同实施例2。
[0066]
溶出度考察结果如下表所示:
[0067]
表3 api粒径范围研究试制样品溶出度检测结果(ph4.5)
[0068][0069][0070]
结论:当api粒径范围在20μm~200μm范围内时,自制品与参比制剂3在ph4.5介质
中均快速溶出(15min溶出度均大于85%),溶出行为相似,故本发明api粒径范围确定为20μm~200μm。
[0071]
实施例4
[0072]
本实施例以溶出度为指标,考察了不同的总混时间。配料处方同实施例1表1中第3组的处方,总混时间如表4,其余的具体的制备工艺同实施例2。考察结果如下表所示:
[0073]
表4总混考察试制样品溶出曲线检测结果(ph4.5)
[0074][0075]
结论:总混时间在5min~10min范围内时,自制品与参比制剂3在ph4.5介质中均快速溶出(15min溶出度均大于85%),溶出行为相似,表明总混时间在5min~10min范围内满足要求。
[0076]
实施例5
[0077]
本实施例考察填充剂微晶纤维素水分含量对溶出及有关物质的影响。配料处方同实施例1表1中第3组的处方,微晶纤维素的含水量如表5,其余的具体的制备工艺同实施例2。考察结果如下表所示:
[0078]
表5微晶纤维素不同水分含量试制样品溶出曲线检测结果(ph4.5)
[0079][0080][0081]
表6微晶纤维素不同水分含量对试制样品有关物质的影响
[0082][0083]
结论:微晶纤维素水分含量在3.9%以下时,自制品与参比制剂3在ph4.5介质中均快速溶出(15min溶出度均大于85%),溶出行为相似,同时微晶纤维素水分含量在3.9%以下时,自制品在高温10天(60℃)条件下杂质增长显著低于参比制剂3,表明微晶纤维素的水分含量在3.9%以下时可更好的确保产品质量。
[0084]
实施例6
[0085]
本实施例考察崩解剂交联羧甲纤维素钠水分含量对溶出及有关物质的影响。配料处方同实施例1表1中第3组的处方,交联羧甲纤维素钠的含水量如表7,其余具体的制备工艺同实施例2。考察结果如下表所示:
[0086]
表7交联羧甲纤维素钠不同水分含量试制样品溶出曲线检测结果(ph4.5)
[0087][0088][0089]
表8交联羧甲纤维素钠不同水分含量对试制样品有关物质的影响
[0090][0091]
结论:交联羧甲纤维素钠水分含量在3.4%以下时,自制品与参比制剂3在ph4.5介质中均快速溶出(15min溶出度均大于85%),溶出行为相似,同时交联羧甲纤维素钠水分含量在3.4%以下时,自制品在高温10天(60℃)条件下杂质增长显著低于参比制剂3,表明崩解剂交联羧甲纤维素钠的水分含量在3.4%以下时可更好的确保产品质量。
[0092]
实施例7
[0093]
本实施例考察压片硬度对溶出的影响。配料处方同实施例1表1中第3组的处方,压片硬度如表9,其余具体的制备工艺同实施例2。考察结果如下表所示:
[0094]
表9不同压片硬度试制样品溶出曲线检测结果(ph4.5)
[0095][0096][0097]
结论:控制压片硬度在50n/mm2~140n/mm2范围时,自制品与参比制剂3在ph4.5介质中均快速溶出(15min溶出度均大于85%),溶出行为相似,表明压片硬度在50n/mm2~140n/mm2范围时满足要求,为进一步保证产品质量,优选压片硬度范围80n/mm2~110n/mm2。
[0098]
实施例8
[0099]
根据实施1表1中的第3组配方,按实施例2的工艺连续制备三批样品(编号分别为3-1、3-2、3-3),考察批间差异性,并与参比品比较。
[0100]
1.自研品及参比品含量均匀度考察结果如下表所示:
[0101]
表10自研品及参比品含量均匀度考察结果
[0102][0103]
结论:三批自研品含量rsd及a 2.2s均明显小于参比品,表明自研品含量均匀度显著优于参比品。
[0104]
2.自研品及参比品溶出度批间差异考察结果如下表所示,自研品及参比品溶出曲线批间差异对比图如附图3所示。
[0105]
表11自研品及参比品批间差异对比数据表(ph4.5,n=6)
[0106][0107]
结论:本发明的三批样品批间rsd明显小于三批参比制剂批间rsd,表明本发明样品溶出批间差异显著小于参比制剂。
[0108]
3.自研品及参比品溶出度在体外不同介质的溶出曲线考察结果如下表所示,溶出曲线图见附图4-1~附图4-4。
[0109]
表12自制品与原研品体外溶出行为比较结果(n=6)
[0110][0111][0112]
结果表明,本发明的三批样品其体外溶出行为与原研一致。
[0113]
4.自研品/原研品质量一致性考察
[0114]
对编号3-1的自研品与原研品(参比制剂3)进行0天、及高温(60℃)放置10天后的质量进行考察,结果如下表所示:
[0115]
表13自制品与原研品质量比较结果
[0116]
[0117]
结论:本发明的三批样品批间rsd明显小于三批参比制剂批间rsd,表明本发明样品溶出批间差异显著小于参比制剂。
[0118]
本发明公开和提出的富马酸丙酚替诺福韦片及其制备方法,本领域技术人员可通过借鉴本文内容,适当改变原料、工艺参数等环节实现。本发明的方法与产品已通过较佳实施例子进行了描述,相关技术人员明显能在不脱离本发明内容、精神和范围内对本文所述的方法和产品进行改动或适当变更与组合,来实现本发明技术。特别需要指出的是,所有相类似的替换和改动对本领域技术人员来说是显而易见的,他们都被视为包括在本发明精神、范围和内容中。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。