1.本发明属于精细化工技术领域,具体涉及一种用于农残检测的衍生化试剂及其制备方法和应用。
背景技术:
2.随着社会的发展和安全意识的提高,人们对药物残留的检测要求也越来越高,所需检测的项目也越来越多。部分药物由于分子结构特点,无法在常规设备中较好地分离,或在常规设备中响应值较低。因此,本领域技术人员常用需要将此类药物进行衍生化处理。样品的衍生化作用主要是把难于分析的物质转化为易于分析的物质,便于量化或分离。一般化学衍生化主要有以下目的:提高样品检测的灵敏度,改善样品混合物的分离度,适合于进一步做结构鉴定等。
3.衍生化试剂需要满足以下使用要求:反应条件温和、迅速、反应彻底,衍生化产物只有一种,副反应及过量衍生化试剂不影响待测物质的分离和检测。
4.草甘膦是一种有机膦类除草剂,是一种非选择性内吸传导型广谱灭生性除草剂,其化学名为n-膦羧甲基甘氨酸(n-phosphonomethyl-glycine,pmg)。草甘膦及其代谢物氨甲基膦酸(aminomethylphosphonic acid,ampa)在植物中的残留已经引起世界的广泛关注。草甘膦的分子结构简单,分子量低,无发色基团,溶于水,不溶于有机溶剂,极性强。
5.公开号为cn201810281233的中国发明专利公开了一种检测茶叶中草铵膦和草甘膦药物残留量的方法。发明在硼酸盐缓冲溶液环境下以fmoc-cl(9-芴基甲基三氯甲烷)为衍生化试剂,在37℃下衍生化10-16小时后离心,过滤后用液相色谱-质谱联用仪测定。该方法中衍生化溶液直接进入质谱,其中的硼酸盐缓冲溶液为无机盐,在质谱中无法分解挥发,将会严重污染离子源甚至质谱内部四级杆等,对质谱将产生极强的抑制作用。
6.gb/t 23750-2009使用水提取,七氟丁醇和三氟乙酸酐作为衍生化试剂,在90℃进行衍生化反应,用气相色谱-质谱联用仪测定粮谷、水果等植物产品中草甘膦及其降解产物氨甲基膦酸残留。而该标准中使用远高于三氟乙酸酐沸点的90℃衍生1h,这将使三氟乙酸酐处于沸腾状态,有一定的泄露风险,同时也可能因为泄露造成衍生化不完全的情况。
7.使用常规的衍生化试剂,无法完全去除杂质,尤其是基质复杂的样品,测试一定数量后,质谱的离子源处均存在大量灰白色至绿色残渣,初步分析为衍生化时的缓冲液及样品中的有机杂质在提取和净化步骤中未去除彻底,这些组分的存在可能污染质谱设备,严重降低质谱设备的响应值。因此,研发新的衍生化试剂进一步净化上机液很有必要。
技术实现要素:
8.本发明针对现有技术存在的问题,提供了一种用于农残检测的衍生化试剂及其制备方法和应用,该方法制得的衍生化试剂得率高、纯度高,在草甘膦检测中的响应值高、稳定性好、准确度高、得到的杂质少、响应迅速且不影响设备的性能。
9.为实现上述目的,本发明采用的技术方案如下:
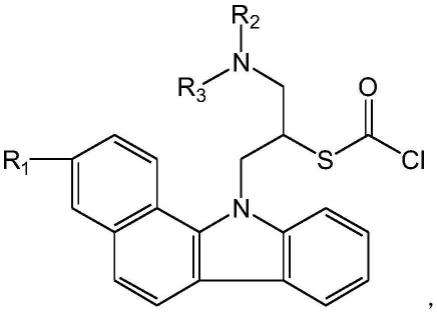
10.一方面,本发明提供一种如下式所示的衍生化试剂:
[0011][0012]
其中,r1,r2,r3各自独立地选自直链烷基。
[0013]
优选地,r1选自c
1-18
直链烷基,r2、r3各自独立地选自c
1-12
直链烷基。
[0014]
进一步优选地,r1选自c
8-18
直链烷基,r2、r3各自独立地选自c
2-10
直链烷基。
[0015]
进一步优选地,r1选自c
10-18
直链烷基,r2、r3各自独立地选自c
2-9
直链烷基。
[0016]
进一步优选地,r1选自c
12-18
直链烷基,r2、r3各自独立地选自c
2-8
直链烷基。
[0017]
进一步优选地,所述衍生化试剂选自
[0018][0018]
中的至少一种。
[0019]
进一步优选地,所述衍生化试剂选自
[0020][0020]
中的至少一种。
[0021]
最优选地,所述衍生化试剂为
[0022]
另一方面,本发明提供上述衍生化试剂的制备方法,包括以下步骤:
[0023]
(1)长链烷基苯和1,4-丁内酯在催化剂条件下进行friedel-crafts酰基化反应,得到长链烷基苯并环己酮;
[0024]
(2)长链烷基苯并环己酮与苯肼在催化剂条件下反应,生成长链烷基氢化苯并咔唑,长链烷基氢化苯并咔唑在还原剂存在下反应,生成长链烷基苯并咔唑;
[0025]
(3)在碱性条件下,长链烷基苯并咔唑与环氧氯丙烷反应,然后与硫脲和相转移催化剂混合反应,生成长链烷基苯并咔唑甲基环硫乙烷;
[0026]
(4)在催化剂的存在下,二烷基胺与长链烷基苯并咔唑甲基环硫乙烷反应后,与三光气和n-甲基咪唑混合,反应得到衍生化试剂。
[0027]
优选地,步骤(1)中,所述长链烷基苯选自十二烷基苯、十四烷基苯、十六烷基苯、十八烷基苯中的至少一种。
[0028]
进一步优选地,所述长链烷基苯选自十六烷基苯、十八烷基苯中的至少一种。
[0029]
更进一步优选地,所述长链烷基苯为十八烷基苯。
[0030]
优选地,步骤(1)中,所述催化剂选自无水氯化铝、氯化锌、三氯化铁、三氟化硼、四氯化锡、硫酸中的至少一种。
[0031]
进一步优选地,所述催化剂选自无水氯化铝、氯化锌、三氯化铁中的至少一种。
[0032]
更进一步优选地,所述催化剂为无水氯化铝。
[0033]
优选地,步骤(1)中,所述长链烷基苯和1,4-丁内酯的摩尔比为1:1.02-1.05,长链烷基苯和催化剂的摩尔比为1:4-5。
[0034]
优选地,所述friedel-crafts酰基化反应具体为:长链烷基苯和1,4-丁内酯在0-10℃下加入催化剂,升温至140-160℃反应6-12h,降温至0-10℃,经后处理得到长链烷基苯并环己酮。
[0035]
优选地,步骤(2)中,所述催化剂选自盐酸、氯化锌、三氯化铁、四氯化锡、硫酸、磷酸中的至少一种。
[0036]
进一步优选地,所述催化剂选自盐酸、硫酸、磷酸中的至少一种。
[0037]
更进一步优选地,所述催化剂为盐酸。
[0038]
优选地,步骤(2)中所述还原剂选自1,4-四氯苯醌、硼氢化钠、三乙酰氧基硼氢化钠、硼氢化锂、n,n-二甲氨基硼氢化锂、二异丁基氢化铝中的至少一种。
[0039]
进一步优选地,所述还原剂选自1,4-四氯苯醌、硼氢化钠、二异丁基氢化铝中的至少一种。
[0040]
更进一步优选地,所述还原剂为1,4-四氯苯醌。
[0041]
优选地,步骤(2)中,所述长链烷基苯并环己酮和苯肼的摩尔比为1:2.5-3,所述长链烷基苯并环己酮与催化剂的摩尔比为1:4-5,所述长链烷基苯并环己酮与1,4-四氯苯醌的摩尔比为1:1-1.15。
[0042]
优选地,步骤(2)中,所述生成长链烷基苯并咔唑的反应具体为:催化剂加热至60-80℃,与苯肼混合升温至80-100℃反应1-2h后,与长链烷基苯并环己酮混合,沸腾反应3-6h;降温至20-30℃,加入二甲苯,向有机层加入还原剂,135-140℃反应3-5h,经后处理得长链烷基苯并咔唑。
[0043]
优选地,步骤(3)中,所述碱性条件的碱选自氢氧化钠、氢氧化钾中的至少一种。
[0044]
优选地,步骤(3)中,所述催化剂选自β-环糊精、四丁基硫酸氢铵中的至少一种。
[0045]
进一步优选地,所述催化剂为β-环糊精。
[0046]
优选地,步骤(3)中所述长链烷基苯并咔唑与环氧氯丙烷反应的摩尔比为1:2-3,所述长链烷基苯并咔唑与碱的摩尔比为1:1-2,所述长链烷基苯并咔唑与硫脲的摩尔比为1:8-12,所述长链烷基苯并咔唑与催化剂的摩尔比为1:1-3。
[0047]
进一步优选地,所述长链烷基苯并咔唑与环氧氯丙烷的摩尔比为1:2.2-3,所述长链烷基苯并咔唑与碱的摩尔比为1:1.2-1.5,所述长链烷基苯并咔唑与硫脲的摩尔比为1:10-12,所述长链烷基苯并咔唑与催化剂的摩尔比为1:1.5-2.5。
[0048]
优选地,步骤(3)中,所述生成长链烷基苯并咔唑甲基环硫乙烷的反应具体为:长链烷基苯并咔唑甲醇溶解后冷却至0-5℃,与20-50%的碱溶液混合,加入环氧氯丙烷,升温至35-50℃,反应3-4小时;有机溶剂萃取后,加入硫脲,催化剂和水,加热至35-50℃,反应7-12h后降温,经后处理得到长链烷基苯并咔唑甲基环硫乙烷。
[0049]
进一步优选地,所述生成长链烷基苯并咔唑甲基环硫乙烷的反应具体为:长链烷基苯并咔唑甲醇溶解后冷却至0-5℃,与30-50%的碱溶液混合,加入环氧氯丙烷,升温至45-50℃,反应3-4小时;冷却至25℃,加入有机溶剂萃取后,调节ph=7-8;加入硫脲,β-环糊精和水,加热至40-50℃,反应8-12h后降温,经后处理得到长链烷基苯并咔唑甲基环硫乙烷。
[0050]
优选地,步骤(4)中,所述二烷基胺选自二乙胺、二正丁胺、二正己胺、二正辛胺中的至少一种。
[0051]
进一步优选地,所述二烷基胺选自二乙胺、二正丁胺、二正己胺中的至少一种。
[0052]
更进一步优选地,所述二烷基胺为二正丁胺。
[0053]
优选地,步骤(4)中所述的催化剂选自n,n-二甲基-4-吡啶胺、三乙胺中的至少一种。
[0054]
进一步优选地,所述的催化剂选自n,n-二甲基-4-吡啶胺。
[0055]
优选地,步骤(4)中,所述长链烷基苯并咔唑甲基环硫乙烷与催化剂的摩尔比为1:0.01-0.02,所述长链烷基苯并咔唑甲基环硫乙烷与二烷基胺的摩尔比为1:1-1.05,所述长链烷基苯并咔唑甲基环硫乙烷与三光气的摩尔比为1:0.4-0.6,所述长链烷基苯并咔唑甲基环硫乙烷与n-甲基咪唑的摩尔比为1:0.04-0.06。
[0056]
优选地,步骤(4)中,所述得到衍生化试剂产物的反应具体为:长链烷基苯并咔唑甲基环硫乙烷与二氯甲烷混合,并降温至0-5℃,与二烷基胺和n,n-二甲基-4-吡啶胺混合,升温至30-50℃反应3-5h,降温至0-10℃;与三光气和n-甲基咪唑混合;在0-5℃下反应4小时升温至30-35℃,继续反应3-6小时,得到衍生化试剂产物。
[0057]
最后,本发明提供上述衍生化试剂在检测草甘膦及其代谢产物的应用。
[0058]
相对于现有技术,本发明具有以下有益效果:
[0059]
(1)本发明的衍生化试剂含有甲硫羰基氯官能团,在一定ph条件时,常温或稍微加热即可迅速与伯胺和仲胺反应,且不受衍生溶液中水的影响;相比常规甲氧羰基氯官能团,甲硫羰基氯与胺基的反应活性更高,反应条件温和,在常温条件下所需的衍生化时间更短;
[0060]
(2)本发明的衍生化试剂含有长链烷基,可增加衍生产物在c18柱中的保留,在纯化和分离步骤可使用70%乙腈水溶液淋洗除去杂质而不影响目标物的保留,使进样溶液杂质更少,色谱分离柱内可能保留的杂质更少,延长色谱分离柱的使用寿命,降低基质效应,提高质谱的相应值,延缓质谱维护周期;
[0061]
(3)本发明的衍生化试剂分子量较高,而杂质一般分子量较小,由于质谱是以质荷比区分各物质的,因此在质谱内可高效地与其他小分子杂质分离。
附图说明
[0062]
图1是本发明实施例1所制得的衍生化试剂a的结构1h nmr图谱;
[0063]
图2是本发明实施例1所制得的衍生化试剂a的红外光谱图;
[0064]
图3是本发明实施例1所制得的衍生化试剂a检测草甘膦的多反应监测色谱图;
[0065]
图4是本发明实施例1所制得的衍生化试剂a检测氨甲基膦酸的多反应监测色谱图。
具体实施方式
[0066]
以下非限制性实施例可以使本领域的普通技术人员更全面的理解本发明,但不以任何方式限制本发明。下述内容仅仅是对本技术要求保护的范围的示例性说明,本领域技术人员可以根据所公开的内容对本技术的发明做出多种改变和修饰,而其也应当属于本技术要求保护的范围之中。
[0067]
下面以具体实施例的方式对本发明作进一步的说明。本发明实施例中所使用的各种化学试剂如无特殊说明均通过常规商业途径获得。
[0068]
下述实施例中,所述十二烷基苯购自上海阿拉丁生化科技股份有限公司,货号为p106336;十六烷基苯购自上海阿拉丁生化科技股份有限公司,货号为h157146;十八烷基苯购自上海阿拉丁生化科技股份有限公司,货号为o159988;β-环糊精购自上海阿拉丁生化科技股份有限公司,货号为c104384;无水三氯化铝购自上海阿拉丁生化科技股份有限公司,货号为a104930;1,4-丁内酯购自济南振达化工有限公司,纯度99.9%;苯肼购自上海阿拉丁生化科技股份有限公司,货号为p108563;1,4-四氯苯醌购自上海阿拉丁生化科技股份有限公司,货号为t105285;环氧氯丙烷购自上海阿拉丁生化科技股份有限公司,货号为e108182;硫脲购自上海阿拉丁生化科技股份有限公司,货号为t112512;二乙胺购自上海阿拉丁生化科技股份有限公司,货号为d110466;二正丁胺购自上海阿拉丁生化科技股份有限公司,货号为d111219;二正辛胺购自上海阿拉丁生化科技股份有限公司,货号为d108994;n,n-二甲基-4-吡啶胺购自上海阿拉丁生化科技股份有限公司,货号为d109207;三光气购自上海阿拉丁生化科技股份有限公司,货号为t103041;n-甲基咪唑购自上海阿拉丁生化科技股份有限公司,货号为m109227。
[0069]
收率计算方法:长链烷基苯并环己酮收率=(长链烷基苯并环己酮重量/理论重量)*100%;长链烷基苯并咔唑收率=(长链烷基苯并咔唑重量/理论重量)*100%;长链烷基苯并咔唑甲基环硫乙烷收率=(长链烷基苯并咔唑甲基环硫乙烷重量/理论重量)*100%;衍生化试剂收率=(衍生化试剂实际重量/理论重量)*100%。
[0070]
纯度的检测方法:液相色谱法。具体检测步骤描述如下:(1)准确称取0.1000g待测化合物溶于无水乙腈,定容至100ml,摇匀;准确量取1.00ml上述溶液,定量至100ml,得到10mg/l待测化合物溶液。(2)准确称取5g硼酸钠溶于300ml水中,再加入700ml乙腈,以70%乙腈水定容,混合均匀后超声除去气泡,以0.22μm膜过滤后,作为流动相使用。(3)液相色谱条件:色谱柱:c18柱,300mm*4,6mm(内径),粒度5μm;流动相流速:1ml/min;检测器:紫外检测器;检测波长:274nm;进样量:10μl。纯度计算方法:面积归一化法。
[0071]
实施例1
[0072]
衍生化试剂a,结构式如下:
[0073][0074]
衍生化试剂a由以下方法制得:
[0075]
(1)在四口瓶中加入38g十八烷基苯和10.3g1,4-丁内酯,在搅拌条件下冷却至5℃,分批次加入64g无水三氯化铝,升温至150℃,反应10小时;降温至5℃,缓慢滴加0℃的1mol/l盐酸溶液500ml,保持温度不超过10℃,滴加完毕后搅拌1小时;加入甲苯100ml,静置
分离出上层有机相,再以1000ml去离子水洗涤有机相2次,分离收集有机相;将有机相过硅胶柱,以甲苯淋洗,15%二氯甲烷-甲苯洗脱,减压蒸馏除去溶剂,得红色油状液体,十八烷基苯并环己酮43.8g,收率为95.60%,纯度为96.43%;
[0076]
(2)在四口瓶中加入480ml1mol/l盐酸,加热至75℃后缓慢加入27g苯肼,升温至100℃回流1小时,缓慢滴加40g十八烷基苯并环己酮,沸腾反应4小时;降温至20℃,加入二甲苯,水洗至水层ph=7,分离并以无水氯化钙干燥有机层;向有机层加入22.5g1,4-四氯苯醌,升温至130℃,反应3小时;降至室温,加入1mol/lnaoh至体系ph=9,静置分离出有机相,减压蒸馏除去二甲苯,以甲醇重结晶,得浅黄色晶体,十八烷基苯并咔唑32.5g,收率为68.97%,纯度为97.55%;
[0077]
(3)将20g十八烷基苯并咔唑,甲醇100ml,搅拌溶解后冷却至4℃,加入4ml40%naoh,缓慢滴加9.2g环氧氯丙烷,升温至50℃,反应3小时;减压蒸馏除去溶剂及过量的环氧氯丙烷后冷却至25℃,加入100ml1,2-二氯乙烷溶解,除去水相,并以去离子水洗涤有机相至ph=7;向有机相中加入由38g硫脲,114gβ-环糊精及500ml水组成的混合物溶液,加热至50℃,反应8小时后降温,弃去水相;将有机相过硅胶柱,以200ml二氯乙烷淋洗,200ml15%丙酮-二氯乙烷洗脱;将洗脱液于45℃下减压蒸馏挥去溶剂,得浅黄色晶体十八烷基苯并咔唑甲基环硫乙烷18.7g,收率为81.07%,纯度为98.72%;
[0078]
(4)取17g十八烷基苯并咔唑甲基环硫乙烷,用50ml二氯甲烷溶解并降温至至4℃,迅速加入4.0g二正丁胺和0.036g催化剂n,n-二甲基-4-吡啶胺,氮气氛中升温至40℃回流反应3小时后,降温至0℃;加入6g三光气和0.015gn-甲基咪唑。在4℃下搅拌4小时后缓慢升温至35℃,继续反应6小时。减压蒸馏至溶剂完全挥发;以无水乙醚重结晶二次,得到浅黄色晶体21.4g,即为衍生化试剂a,收率为92.96%,衍生化试剂a的总收率为49.69%,纯度为98.22%。
[0079]
对获得的衍生化试剂a进行1h nmr和红外光谱检测:
[0080]
衍生化试剂a的核磁谱图如图1所示,(1)0.00—定位化合物tms的h化学位移;(2)0.87—ch3的h化学位移,峰面积152.5,9h;(3)1.28—烷基ch2(不与苯环、n连接的ch2)的h化学位移,峰面积573.4,34h;(4)2.36—与叔胺n连接的烷基ch2的h化学位移,峰面积69.5,4h;(5)1.39—与(3)相邻的ch2的h化学位移,峰面积68.9,4h;(6)2.62—与苯环连接的ch2的h化学位移,峰面积33.6,2h;(7)1.62—与(5)相邻的ch2的h化学位移,峰面积33.9,2h;(8)3.00—与叔胺的n相邻的ch2及s相邻的ch2的h的化学位移,峰面积50.2,3h;(9)4.48—与咔唑的n相邻的ch2的h的化学位移,峰面积33.9,2h;(10)7.16—芳环上的h的化学位移,峰面积101.4,6h;(11)7.46—芳环上的h的化学位移,峰面积16.9,1h;(12)7.55—芳环上的h的化学位移,峰面积17.2,1h;(13)7.66—芳环上的h的化学位移,峰面积16.7,1h。
[0081]
衍生化试剂a的红外光谱图如图2所示,其在3031、1600、1498、1030、769、740、690cm-1
处出现苯基、联苯基特征吸收峰,在1769cm-1
处出现酰氯基特征吸收峰,在723cm-1
处出现碳数≥4的直链烃基特征吸收峰,在1438cm-1
处出现c-s-c=o特征吸收峰,在1143、1066cm-1
处出现咔唑的c-n-c特征吸收峰,在1357、1247cm-1
处出现叔胺的c-n-c特征吸收峰,在2956、1463cm-1
处出现ch3的特征吸收峰,在2925、2826cm-1
处出现ch2特征吸收峰。证明衍生化试剂a制备成功。
[0082]
实施例2
[0083]
衍生化试剂b,结构式如下:
[0084][0085]
衍生化试剂b由以下方法制得:
[0086]
与实施例1不同的是,步骤(1)中的十八烷基苯替换为十二烷基苯,其余配比和反应条件皆相同,得红色油状液体,十二烷基苯并环己酮46.72g,收率为96.39%,纯度为95.01%;步骤(2)与实施例1相同,得十二烷基苯并咔唑35.28g,收率为71.96%,纯度为96.34%;步骤(3)与实施例1相同,得十二烷基苯并咔唑甲基环硫乙烷20.01g,收率为84.32%,纯度为95.93%;
[0087]
与实施例1不同的是,步骤(4)中的二正丁烷替换为二正辛胺,其余配比和反应条件皆相同,得衍生化试剂b26.75g,收率为94.57%,衍生化试剂b的总收率为55.31%,纯度为97.34%。
[0088]
实施例3
[0089]
衍生化试剂c,结构式如下:
[0090][0091]
衍生化试剂c由以下方法制得:
[0092]
步骤(1)-(3)与实施例1相同,与实施例1不同的是,步骤(4)中的二正丁胺替换为二乙胺,其余配比和反应条件皆相同,得衍生化试剂c20.38g,总收率为52.82%,纯度为97.99%。
[0093]
实施例4
[0094]
衍生化试剂d,结构式如下:
[0095][0096]
衍生化试剂d由以下方法制得:
[0097]
与实施例1不同的是,步骤(1)中的十八烷基苯替换为十六烷基苯,其余配比和反应条件皆相同,得十六烷基苯并环己酮44.37g,收率为95.34%,纯度为97.11%;
[0098]
步骤(2)与实施例1相同,得十六烷基苯并咔唑34.41g,收率为72.19%,纯度为96.89%;
[0099]
步骤(3)与实施例1相同,得十六烷基苯并咔唑甲基环硫乙烷19.74g,收率为84.86%,纯度为95.73%;
[0100]
步骤(4)与实施例1相同,得衍生化试剂d24.09g,收率为95.61%,衍生化试剂d的总收率为55.84%,纯度为97.91%。
[0101]
实验检测:检测草甘膦及其代谢产物
[0102]
实验方法1:实施1所得的衍生化试剂a以及常规衍生化试剂fmoc-cl分别作为衍生化试剂,依照下列方法进行草甘膦及其代谢产物的检测。
[0103]
(1)提取:称取1.0g红茶(已验证不含草甘膦及其代谢物氨甲基膦酸的空白基质样品),添加不同浓度的草甘膦和氨甲基膦酸标准溶液,以10ml0.5%氨水涡旋混匀,超声提取30min后,4000rpm离心5min,将上清液分别转入25ml玻璃比色管,待衍生;
[0104]
(2)衍生化:准确称取0.5g衍生化试剂溶于100ml无水乙腈,超声溶解后摇匀。取5ml硼酸盐缓冲液(5%,ph=9.0),加入步骤(1)的待衍生溶液中,加入5ml乙腈和2ml衍生化溶液后密封混匀,室温中振荡反应2小时后待纯化;
[0105]
(3)纯化:将上清液通过预先以3ml甲醇,3ml水活化的c18固相萃取小柱,弃去流出液。依次以5ml去离子水,5ml70%乙腈水淋洗,弃去淋洗液,5ml乙腈洗脱,将洗脱液在35℃下氮吹至近干,加入1.0ml流动相,涡旋混合30秒后,以0.22μm滤膜过滤除去杂质,得到无色透明溶液,待仪器测定;
[0106]
(4)草甘膦及其代谢物氨甲基膦酸浓度和回收率的检测:
[0107]
液相色谱条件:色谱柱:waters acquity uplc beh色谱柱(1.7μm,50mm*1.0mm),柱温30℃,流速400μl/min,进样量5μl,梯度洗脱程序见表1;
[0108]
表1
[0109][0110][0111]
质谱条件:waters三重四极杆串联质谱联用仪uplc-xevo tq-s micro,电喷雾esi 离子源;扫描方式:多反应监测(mrm);离子源温度490℃;定性离子对,定量离子对,去簇电压和碰撞气能量质谱参数见表2及表3。
[0112]
表2
[0113][0114]
表3
[0115][0116]
检测结果见图3,图4和表5。
[0117]
实验方法2:采用sn/t 1923-2007的检测方法,以常规衍生化试剂fmoc-cl作为衍生化试剂,对草甘膦及其代谢产物进行检测:
[0118]
(1)提取:称取5g红茶(已验证不含草甘膦及其代谢物氨甲基膦酸的空白基质样品)于250ml塑料离心管中,加入草甘膦内标工作溶液,加入100ml水(茶叶样品浸泡0.5h)和50ml二氯甲烷,振荡20min,于4000r/min离心10min。将上层水溶液转移至另一塑料离心管
中,残渣再加入50ml水重复提取一次,合并上层水溶液,充分混匀后,取4.5ml至10ml塑料试管中,加0.5ml酸度调节剂,混匀;
[0119]
(2)净化:cax小柱经10ml水活化后,加1.0ml提取液,用0.7mlcax洗脱液淋洗两次,再用11ml洗脱液洗脱并收集,洗脱液于45℃减压旋转蒸发至干,加1ml5%硼酸盐缓冲溶液溶解残渣,此时ph约为9左右,需要时用20%氢氧化钠溶液和3mol/l盐酸溶液、0.3mol/l盐酸溶液调节ph至9;
[0120]
(3)衍生化:取混合标准工作溶液各1.0ml加入200μl5%喷酸盐缓冲溶液,混匀。此系列溶液和净化后样液分别加入200μlfmoc-cl丙酮溶液,混匀,室温下进行衍生化反应,放置过夜。将衍生化后溶液通过0.45μm滤膜,供液相色谱-串联质谱测定。
[0121]
(4)草甘膦及其代谢物氨甲基膦酸浓度和回收率的检测:液相色谱条件:色谱柱:waters acquity uplc beh色谱柱(1.7μm,50mm*1.0mm),柱温30℃,流速400μl/min,进样量5μl,梯度洗脱程序见表4;
[0122]
表4
[0123]
时间,min0.1%甲酸乙腈溶液,%5mmol/l乙酸铵 0.2%甲酸溶液,%0.0020.080.01.5070.030.02.5095.05.03.5095.05.04.0020.080.06.0020.080.0
[0124]
质谱条件:waters三重四极杆串联质谱联用仪uplc-xevo tq-s micro,电喷雾esi 离子源;扫描方式:多反应监测(mrm);离子源温度490℃;定性离子对,定量离子对,去簇电压和碰撞气能量质谱参数见表3。
[0125]
检测结果见表5。
[0126]
实验结果:
[0127]
从图3和图4中可以看出,将本发明的衍生化试剂用于农残的检测,可以有效的检测到草甘膦及其代谢物氨甲基膦酸。说明,本发明的衍生化试剂在草甘膦及其代谢物检测中具有很好的应用效果,所使用的的衍生化试剂可高效、温和且快速的对草甘膦及其代谢物氨甲基膦酸进行反应,从而加快反应进程。
[0128]
从表5中可以看出,添加不同浓度草甘膦及其代谢物氨甲基膦酸的茶叶以实验方法1进行检测,经过本发明所述的衍生化试剂a作为衍生化试剂进行衍生化后纯化上机检测,草甘膦及其代谢物的回收率及实验稳定性都很好,所得结果符合残留检测要求。而使用fmoc-cl作为衍生化试剂进行相同步骤纯化,在淋洗的过程中,大量衍生化产物被高浓度乙腈水洗脱,进入废液,其回收率极低,不符合残留检测要求。作为对比,对添加了不同浓度草甘膦及其代谢物氨甲基膦酸的茶叶以实验方法2进行检测,基本复合残留检测要求,但其回收率比使用本发明所述衍生化试剂a作为衍生化试剂的方法低。综上所述,本发明所述的采用衍生化试剂进行草甘膦及其代谢物氨甲基膦酸生物检测,是一种高效、准确、绿色、无污染的方法。本发明的衍生化试剂可用于草甘膦及其代谢物氨甲基膦酸的残留分析检测。
[0129]
表5
[0130][0131]
最后应当说明的是,以上内容仅用以说明本发明的技术方案,而非对本发明保护范围的限制,本领域的普通技术人员对本发明的技术方案进行的简单修改或者等同替换,均不脱离本发明技术方案的实质和范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
