1.本发明属于化合物合成技术领域,尤其涉及一种以二苯并环辛炔-四聚乙二醇-活性酯(dbco-peg4-nhs ester,化合物11)为代表的一类化合物、制备及应用鉴定方法。
背景技术:
2.目前,环辛炔醇、bcn、dbco-cooh和dbco-nhs等试剂的成本仍超过1000美元/g,这对于聚合物或材料化学来说是不可接受的高成本。虽然已经开展了工作(wo2018089373),以开发有效的可扩展的方法,较少反应性的应变环辛烷,如(2-环辛烷-1-乙氧基)乙酸,目前发表的合成方法,均需要经过色谱纯化;同时所需材料多,成本高且制备规模小,收率不高。
3.通过上述分析,现有技术存在的问题及缺陷为:现有文献(j.org.chem.2010,75,627)需要的二异丙基铝(dibal-h,6~7当量,一步从化合物2合成化合物4,收率73%)而所用的二异丙基铝非常容易燃烧,且之后还要用大量氟化钠处理,步骤繁琐,制备规模在小(仅克级),如大量制备需要特殊的设备。从化合物4到化合物6的制备,专利(wo2018089373)使用的试剂是(琥玻酸酐),从而限制了此处碳链的长度和变化。
4.二苯并环辛炔-四聚乙二醇-活性酯(11)是2013年以来,当前研究和开发靶向药物的重要链接剂,用于分子荧光探针(angew.chem.int.ed.2013,52,7756;angew.chem.int.ed.,2015,54,743;cn104749369;nano lett.2015,15,8217;bioconjugate chem.2017,28,1151;langmuir,2020,36,4272-4279)。本发明之优点在于:1、由两步反应,从化合物2重排到化合物3再还原到化合物4,巧妙的避免使用非常容易燃烧的二异丙基铝(dibal-h)和后处理的大量氟化钠。实现了少纯化,比原文献更高收率(~96%),理论上可以到百克级,甚至公斤级,并大大降低了低成本也减低了反应的危险性。
5.2、在从化合物4到化合物6的合成中,使用代表性的丁二酸单乙酯酰氯,不仅可以任意控制此处碳链的长短(如通式1和通式2所示),同时大大提高了此类试剂的灵活性,以适应不同生化条件的需要,而且可以改变其它带有官能团的侧链,提高试剂的适用范围。
6.解决上述技术问题后带来的积极效果为:
7.本发明不仅彻底克服了现有文献成本高且制备规模小,收率低问题,而且大大扩展了试剂的灵活性和使用范围,有着广泛的应用前景和不可估量的经济效益
技术实现要素:
8.针对现有技术存在的问题,本发明提供了一种二苯并环辛炔-四聚乙二醇-活性酯类化合物、制备方法及用途。一类以二苯并环辛炔-四聚乙二醇-活性酯(化合物11)为代表的一类化合物。
9.本发明是这样实现的,一种二苯并环辛炔-四聚乙二醇-活性酯类化合物,其结构式为通式为1或通式2:
[0010][0011]
通式1:n=0和正整数;m=正整数;x=o或s或n或ch2或它们的任意组合
[0012][0013]
通式2:n=0和正整数;m=正整数;x=o或s或n或或它们的任意组合。
[0014]
进一步,所述二苯并环辛炔-四聚乙二醇-活性酯结构式为:
[0015][0016]
本发明的另一目的在于提供一种二苯并环辛炔-四聚乙二醇-活性酯类化合物的制备方法反应式为:
[0017]
[0018]
进一步,所述化合物制备方法包括以下步骤:
[0019]
从5-二苯并环庚烯酮(1)经由5h-二苯并[a,d]环庚烯-5-酮肟(2)、二苯并[b,f]吖辛因-6(5h)-酮(3)、(z)-5,6-二氢二苯并[b,f]氮杂辛(4)、(z)-5,6-二氢二苯并[b,f]氮杂辛琥珀酰胺乙酯(5)、(z)-5,6-二氢二苯并[b,f]氮杂辛单琥珀酰胺(6)、5,6-二溴-二氢二苯并[b,f]氮杂辛单琥珀酰胺(7)、二苯并环辛炔-羧基(8)、dbco-peg4-acid叔丁酯(9)、dbco-peg4-acid(10),最后将10与n-羟基琥珀酰亚胺以edci偶联,得到黄色油状物11。本发明只在在必要步才用柱色谱纯化的前提下,简单、高效、高产地合成了高纯度目标化合物11。
[0020]
具体包括:步骤一,基于化合物1分别合成化合物2、化合物3;分别进行化合物4、化合物5、化合物6的合成;将溴素缓慢加到化合物6的二氯甲烷溶液中反应生成化合物7;
[0021]
步骤二,将化合物7与1m叔丁醇钾的四氢呋喃溶液反应生成化合物8;将所述化合物8溶于二氯甲烷,与edci、edci、dmap、15-氨基-4,7,10,13-四氧杂十五烷酸叔丁酯反应生成化合物9;
[0022]
步骤三,于室温下,将化合物9溶于二氯甲烷和三氟乙酸,室温1h,tlc显示反应完全后将反应液拉干过柱子得到黄色油状物即化合物10;于室温下,将化合物10缓慢加到化合物n-羟基琥珀酰亚胺和edci的二氯甲烷溶液中去,室温过夜后加入200ml水,用二氯甲烷萃取,水洗5次,拉干后得到黄色油状物,即可。
[0023]
进一步,所述于室温下基于化合物1分别合成化合物2、化合物3包括:
[0024]
于室温下,将盐酸羟胺缓慢加到化合物1的吡啶溶液中,120℃回流过夜,当tlc显示化合物1反应完全时,旋除大部分吡啶,倒入1l冰水,加入100ml浓盐酸和200ml正己烷,滤出白色固体,充分干燥,得到化合物2;
[0025]
于室温下,将三氟乙酸加到化合物2中,80℃回流过夜后,tlc显示化合物2反应完全时,旋干三氟乙酸,加入500ml水,过滤得到的固体依次用200ml dcm和100ml乙醇打浆,过滤得到灰色固体即为化合物3。
[0026]
进一步,所述于0℃下,分别进行化合物4、化合物5、化合物6的合成包括:
[0027]
于0℃下,将四氢锂铝缓慢加到化合物3的200ml的乙醚溶液中去,回流过夜,tlc显示反应完全时,反应液加水淬灭,过滤得到黄色固体即化合物4;
[0028]
于0℃下,将丁二酸单乙酯酰氯缓慢加到化合物4和三乙胺的二氯甲烷溶液中去,室温1h,tlc显示反应完全后,反应液加入200ml水,二氯甲烷萃取,水洗3次,拉干过柱子得到白色固体即为化合物5;
[0029]
于0℃下,将氢氧化钠缓慢加到化合物5的20ml四氢呋喃、20ml甲醇和20ml水中去,室温2h,tlc显示反应完全后,旋除反应液中的甲醇和四氢呋喃,调酸后加入20ml水,二氯甲烷萃取,水洗3次,拉干即可得化合物6。
[0030]
进一步,所述于10℃下,将溴素缓慢加到化合物6的二氯甲烷溶液中反应生成化合物7包括:
[0031]
将溴素缓慢加到化合物6的二氯甲烷溶液中,室温反应2h,tlc显示反应完全后向反应液加入200ml水,用硫代硫酸钠淬灭溴素,二氯甲烷萃取,水洗3次,拉干过柱子得到白色固体,用600ml乙酸乙酯打浆得到白色固体即化合物7。
[0032]
进一步,步骤二中所述将1.3l 1m叔丁醇钾的四氢呋喃溶液缓慢加到化合物7的四
氢呋喃溶液中去包括:分批次加入,于3h内加全部加入。
[0033]
进一步,步骤五中,所述,将化合物8溶于二氯甲烷,与edci、edci、dmap、15-氨基-4,7,10,13-四氧杂十五烷酸叔丁酯反应生成化合物9包括:
[0034]
室温下,将化合物8溶于二氯甲烷,加入edci和dmap,最后加入15-氨基-4,7,10,13-四氧杂十五烷酸叔丁酯,室温过夜,tlc显示反应完全后向反应液加入水,调至酸性,用二氯甲烷萃取,水洗3次,拉干后过柱子得到黄色油状物即为化合物9。
[0035]
进一步,所述化合物制备方法还包括:
[0036]1h nmr(400mhz,dmso)δ11.34(s,1h),7.68
–
7.15(m,8h),6.95(d,j=0.7hz,2h);
[0037]1h nmr(400mhz,dmso)δ7.28
–
7.04(m,4h),6.97
–
6.69(m,2h),6.57
–
6.33(m,3h),6.28
–
6.14(m,2h),4.44(d,j=7.2hz,2h);
[0038]1h nmr(400mhz,cdcl3)δ7.73(d,j=7.7hz,1h),7.30
–
7.00(m,6h),6.90(d,j=7.5hz,1h),5.89(d,j=9.9hz,1h),5.82(d,j=14.9hz,1h),5.16(d,j=9.9hz,1h),4.21(d,j=14.9hz,1h),3.01
–
2.81(m,1h),2.73
–
2.44(m,3h);
[0039]1h nmr(400mhz,cdcl3)δ7.68(d,j=7.5hz,1h),7.50
–
7.18(m,7h),5.16(d,j=13.9hz,1h),3.70(d,j=13.8hz,1h),2.66(dddd,j=46.9,16.9,8.9,5.3hz,2h),2.36(ddd,j=16.9,6.3,5.3hz,1h),2.16
–
1.88(m,1h);
[0040]1h nmr(400mhz,cdcl3)δ7.61(d,j=7.3hz,1h),7.53
–
7.42(m,1h),7.41
–
7.07(m,6h),6.23(t,j=5.3hz,1h),5.08(d,j=13.8hz,1h),3.68
–
3.45(m,14h),3.39(dt,j=6.6,5.1hz,2h),3.28(t,j=5.3hz,2h),2.76(ddd,j=16.7,8.3,6.6hz,1h),2.46
–
2.33(m,3h),2.09(dt,j=15.3,6.2hz,1h),1.87(dt,j=16.8,6.2hz,1h),1.38(s,9h);
[0041]1h nmr(400mhz,cdcl3)δ7.65(d,j=7.4hz,1h),7.56
–
7.43(m,1h),7.43
–
7.11(m,6h),6.30(s,1h),5.13(d,j=13.8hz,1h),3.89
–
3.20(m,19h),2.82(dt,j=23.3,10.6hz,7h),2.54
–
2.33(m,1h),2.14(dt,j=15.1,6.2hz,1h),1.92(dt,j=16.7,6.2hz,1h)。
[0042]
所述基于化合物1合成化合物2,所需的honh2可以为任何可能形式的盐,但不局限于盐及其等价物,如盐酸盐、硫酸盐、碳酸盐等。
[0043]
进一步,所述基于化合物2合成化合物3,所需的酸,不仅限于如图所示的三氟乙酸,也包括盐酸,氢溴酸,硫酸,三氯乙酸,高氯酸以及它们的溶液或者任意一种或几种的组合。
[0044]
所述基于化合物3合成化合物4,所需的还原试剂,不仅限于如图所示的氢化铝锂,也包括硼烷,二异丙基铝,硼氢化钠,以及它们的溶液或者任意一种或几种的组合,或者是与任何金属盐的任意组合。
[0045]
所述基于化合物4合成化合物5,所需的酰化试剂,不仅限于如图所示的丁二酸单乙酯酰氯,也包括丁二酸单烷基酯酰氯,以及丁二酸单烷基酯与氯甲酸乙酯包括氯甲酸烷基酯所形成的混酸酐,(这里的烷基是指:甲级,丙基,异丙基,丁基等)。
[0046]
所述基于化合物5合成化合物6,所需的水解试剂,不仅限于如图所示的氢氧化钠,也包括任何主副族元素的氢氧化物,如lioh,koh,csoh,ca(oh)2,以及任何主副族元素的如k2co3,cs2co3等。
[0047]
所述基于化合物7合成化合物8,所需的脱卤素的试剂,不仅限于如图所示的叔丁醇钾,也包括任何其它的醇钾、钠,如叔丁醇钠,叔戊醇钠,叔戊醇钾,氢化钠,氢化钾等。
[0048]
所述基于化合物8合成化合物9,和化合物10合成化合物11所需的偶联试剂,不仅限于如图所示的edci,也包括任何其它,如dcc等。
[0049]
所述基于化合物9合成化合物10,所需的酸,不仅限于如图所示的三氟乙酸,也包括盐酸,氢溴酸,硫酸,三氯乙酸,高氯酸以及它们的溶液或者任意一种或几种的组合。
[0050]
本发明另一目的在于提供一种所述二苯并环辛炔-四聚乙二醇-活性酯类化合物作为官能团化的聚乙二醇链接剂修饰蛋白质,在制备诊断试剂,adc挂载,脂质体的表面靶向分子挂载上的用途。
[0051]
结合上述的所有技术方案,本发明所具备的优点及积极效果为:
[0052]
本发明从5-二苯并环庚烯酮(1)经由5h-二苯并[a,d]环庚烯-5-酮肟(2)、二苯并[b,f]吖辛因-6(5h)-酮(3)、(z)-5,6-二氢二苯并[b,f]氮杂辛(4)、(z)-5,6-二氢二苯并[b,f]氮杂辛琥珀酰胺乙酯(5)、(z)-5,6-二氢二苯并[b,f]氮杂辛单琥珀酰胺(6)、5,6-二溴-二氢二苯并[b,f]氮杂辛单琥珀酰胺(7)、二苯并环辛炔-羧基(8)、dbco-peg4-acid叔丁酯(9)、dbco-peg4-acid(10),最后将10与n-羟基琥珀酰亚胺以edci偶联,得到黄色油状物11。本发明只在在必要步才用柱色谱纯化的前提下,简单、高效、高产地合成了高纯度目标化合物11。
[0053]
本发明允许每个中间体的简单分离,而不需要任何色谱纯化步骤,且收率优秀至接近定量,实现了无需柱色谱纯化,有些步骤仅需简单的分离,有些则通过重结晶实现,高效、高产、在无需柱色谱纯化的前提下,合成了相应的化合物。
附图说明
[0054]
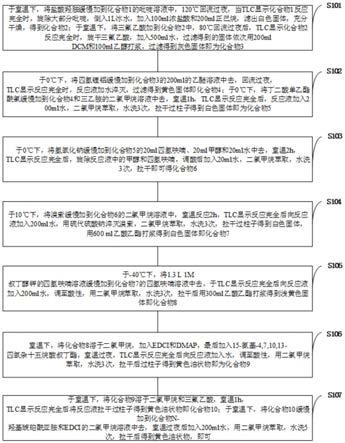
图1是本发明实施例提供的化合物11(二苯并环辛炔-四聚乙二醇-活性酯)制备方法流程图。
具体实施方式
[0055]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0056]
针对现有技术存在的问题,本发明提供了一种化合物及其制备方法,下面结合附图对本发明作详细的描述。
[0057]
本发明提供的一种二苯并环辛炔-四聚乙二醇-活性酯类化合物,其结构式为通式为1或通式2:
[0058][0059]
通式1:n=0和正整数;m=正整数;x=o或s或n或ch2或它们的任意组合
[0060][0061]
通式2:n=0和正整数;m=正整数;x=o或s或n或或它们的任意组合。
[0062]
本发明实施例提供的化合物11如下:
[0063][0064]
本发明实施例提供的化合物11制备方法如下:
[0065][0066]
如图1所示,本发明实施例提供的化合物11制备方法包括以下步骤:
[0067]
s101,于室温下,将盐酸羟胺缓慢加到化合物1的吡啶溶液中,120℃回流过夜,当tlc显示化合物1反应完全时,旋除大部分吡啶,倒入1l冰水,加入100ml浓盐酸和200ml正己烷,滤出白色固体,充分干燥,得到化合物2;于室温下,将三氟乙酸加到化合物2中,80℃回流过夜后,tlc显示化合物2反应完全时,旋干三氟乙酸,加入500ml水,过滤得到的固体依次
用200ml dcm和100ml乙醇打浆,过滤得到灰色固体即为化合物3。
[0068]
s102,于0℃下,将四氢锂铝缓慢加到化合物3的200ml的乙醚溶液中去,回流过夜,tlc显示反应完全时,反应液加水淬灭,过滤得到黄色固体即化合物4;于0℃下,将丁二酸单乙酯酰氯缓慢加到化合物4和三乙胺的二氯甲烷溶液中去,室温1h,tlc显示反应完全后,反应液加入200ml水,二氯甲烷萃取,水洗3次,拉干过柱子得到白色固体即为化合物5;
[0069]
s103,于0℃下,将氢氧化钠缓慢加到化合物5的20ml四氢呋喃、20ml甲醇和20ml水中去,室温2h,tlc显示反应完全后,旋除反应液中的甲醇和四氢呋喃,调酸后加入20ml水,二氯甲烷萃取,水洗3次,拉干即可得化合物6;
[0070]
s104,于10℃下,将溴素缓慢加到化合物6的二氯甲烷溶液中,室温反应2h,tlc显示反应完全后向反应液加入200ml水,用硫代硫酸钠淬灭溴素,二氯甲烷萃取,水洗3次,拉干过柱子得到白色固体,用600ml乙酸乙酯打浆得到白色固体即化合物7;
[0071]
s105,于-40℃下,将1.3l 1m叔丁醇钾的四氢呋喃溶液缓慢加到化合物7的四氢呋喃溶液中去,于tlc显示反应完全后向反应液加入200ml水,调至酸性,用二氯甲烷萃取,水洗3次,拉干后用300ml乙酸乙酯打浆得到浅黄色固体即化合物8;
[0072]
s106,室温下,将化合物8溶于二氯甲烷,加入edci和dmap,最后加入15-氨基-4,7,10,13-四氧杂十五烷酸叔丁酯,室温过夜,tlc显示反应完全后向反应液加入水,调至酸性,用二氯甲烷萃取,水洗3次,拉干后过柱子得到黄色油状物即为化合物9;
[0073]
s107,于室温下,将化合物9溶于二氯甲烷和三氟乙酸,室温1h,tlc显示反应完全后将反应液拉干过柱子得到黄色油状物即化合物10;于室温下,将化合物10缓慢加到化合物n-羟基琥珀酰亚胺和edci的二氯甲烷溶液中去,室温过夜后加入200ml水,用二氯甲烷萃取,水洗5次,拉干后得到黄色油状物,即可。
[0074]
步骤s105中本发明实施例提供的将1.3l 1m叔丁醇钾的四氢呋喃溶液缓慢加到化合物7的四氢呋喃溶液中去包括:分批次加入,于3h内加全部加入。
[0075]
本发明实施例提供的化合物制备方法还包括:
[0076]1h nmr(400mhz,dmso)δ11.34(s,1h),7.68
–
7.15(m,8h),6.95(d,j=0.7hz,2h);
[0077]1h nmr(400mhz,dmso)δ7.28
–
7.04(m,4h),6.97
–
6.69(m,2h),6.57
–
6.33(m,3h),6.28
–
6.14(m,2h),4.44(d,j=7.2hz,2h);
[0078]1h nmr(400mhz,cdcl3)δ7.73(d,j=7.7hz,1h),7.30
–
7.00(m,6h),6.90(d,j=7.5hz,1h),5.89(d,j=9.9hz,1h),5.82(d,j=14.9hz,1h),5.16(d,j=9.9hz,1h),4.21(d,j=14.9hz,1h),3.01
–
2.81(m,1h),2.73
–
2.44(m,3h);
[0079]1h nmr(400mhz,cdcl3)δ7.68(d,j=7.5hz,1h),7.50
–
7.18(m,7h),5.16(d,j=13.9hz,1h),3.70(d,j=13.8hz,1h),2.66(dddd,j=46.9,16.9,8.9,5.3hz,2h),2.36(ddd,j=16.9,6.3,5.3hz,1h),2.16
–
1.88(m,1h);
[0080]1h nmr(400mhz,cdcl3)δ7.61(d,j=7.3hz,1h),7.53
–
7.42(m,1h),7.41
–
7.07(m,6h),6.23(t,j=5.3hz,1h),5.08(d,j=13.8hz,1h),3.68
–
3.45(m,14h),3.39(dt,j=6.6,5.1hz,2h),3.28(t,j=5.3hz,2h),2.76(ddd,j=16.7,8.3,6.6hz,1h),2.46
–
2.33(m,3h),2.09(dt,j=15.3,6.2hz,1h),1.87(dt,j=16.8,6.2hz,1h),1.38(s,9h);
[0081]1h nmr(400mhz,cdcl3)δ7.65(d,j=7.4hz,1h),7.56
–
7.43(m,1h),7.43
–
7.11(m,6h),6.30(s,1h),5.13(d,j=13.8hz,1h),3.89
–
3.20(m,19h),2.82(dt,j=23.3,10.6hz,
7h),2.54
–
2.33(m,1h),2.14(dt,j=15.1,6.2hz,1h),1.92(dt,j=16.7,6.2hz,1h)。
[0082]
下面结合具体实施例对本发明的技术方案做进一步说明。
[0083]
实施例1:
[0084]
化合物2的合成
[0085]
室温下,将盐酸羟胺(92g,1.31mol)缓慢加到化合物1(180g,0.873mol)的吡啶(630ml)溶液中去,加完120℃回流过夜,tlc显示化合物1已无,旋掉大部分吡啶,倒入1l冰水,加入100ml浓盐酸和200ml正己烷,滤出白色固体,充分干燥有200g,收率103.5%,无须进一步纯化直接以100%收率投往下一步。
[0086]
化合物3的合成
[0087]
室温下,将三氟乙酸(90ml)加到化合物2(18.7g,84.6mmol)中,80℃回流过夜后,tlc显示化合物2已无。旋干三氟乙酸,加入500ml水,过滤得到的固体依次用200ml dcm和100ml乙醇打浆,过滤得到16g灰色固体,收率80%。
[0088]1h nmr(400mhz,dmso)δ11.34(s,1h),7.68
–
7.15(m,8h),6.95(d,j=0.7hz,2h).
[0089]
化合物4的合成
[0090]
0℃下,将四氢锂铝(24g,0.63mol)缓慢加到化合物3(14g,63.3mmol)的乙醚(200ml)溶液中去,加完回流过夜,tlc显示反应已完全。反应液加水淬灭,过滤得到12.60g黄色固体,收率96%。
[0091]1h nmr(400mhz,dmso)δ7.28
–
7.04(m,4h),6.97
–
6.69(m,2h),6.57
–
6.33(m,3h),6.28
–
6.14(m,2h),4.44(d,j=7.2hz,2h).
[0092]
化合物5的合成
[0093]
0℃下,将丁二酸单乙酯酰氯(8.73g,53.06mmol)缓慢加到化合物4(10g,48.24mmol)和三乙胺(5.85g,57.89mmol)的二氯甲烷(100ml)溶液中去,加完室温1h,tlc显示反应已完全。反应液加入200ml水,二氯甲烷萃取,水洗3次,拉干过柱子得到7.2g白色固体,收率89%。
[0094]
化合物6的合成
[0095]
0℃下,将氢氧化钠(1.72g,42.93mmol)缓慢加到化合物5(10g,48.24mmol)的四氢呋喃(20ml)、甲醇(20ml)和水(20ml)中去,加完室温2h,tlc显示反应已完全。反应液旋掉甲醇和四氢呋喃,调酸后加入20ml水,二氯甲烷萃取,水洗3次,拉干以100%收率投往下一步。
[0096]
化合物7的合成
[0097]
10℃下,将溴素(58.60g,0.367mol)缓慢加到化合物6(102.48g,0.333mol)的二氯甲烷(1.5l)溶液中去,加完室温2h,tlc显示反应已完全。反应液加入200ml水,用硫代硫酸钠淬灭溴素,二氯甲烷萃取,水洗3次,拉干过柱子得到白色固体,用600ml乙酸乙酯打浆得到129g白色固体,收率80.5%。
[0098]1h nmr(400mhz,cdcl3)δ7.73(d,j=7.7hz,1h),7.30
–
7.00(m,6h),6.90(d,j=7.5hz,1h),5.89(d,j=9.9hz,1h),5.82(d,j=14.9hz,1h),5.16(d,j=9.9hz,1h),4.21(d,j=14.9hz,1h),3.01
–
2.81(m,1h),2.73
–
2.44(m,3h).
[0099]
化合物8的合成
[0100]-40℃下,将1.3l 1m叔丁醇钾的四氢呋喃溶液缓慢加到化合物7(140g,299.6mmol)的四氢呋喃(1.2l)溶液中去,分批控制在3h内加完,tlc显示反应已完全。反应
液加入200ml水,调至酸性,用二氯甲烷萃取,水洗3次,拉干后用300ml乙酸乙酯打浆得到88g浅黄色固体,收率96.7%。
[0101]1h nmr(400mhz,cdcl3)δ7.68(d,j=7.5hz,1h),7.50
–
7.18(m,7h),5.16(d,j=13.9hz,1h),3.70(d,j=13.8hz,1h),2.66(dddd,j=46.9,16.9,8.9,5.3hz,2h),2.36(ddd,j=16.9,6.3,5.3hz,1h),2.16
–
1.88(m,1h).
[0102]
化合物9的合成
[0103]
室温下,将化合物8(88.5g,0.29mol)溶于二氯甲烷(0.9l),加入edci(61.1g,0.319mol)和dmap(3.54g,29mmol),最后加入15-氨基-4,7,10,13-四氧杂十五烷酸叔丁酯(此化合物是从武汉博仁凯润药业有限公司购买,具体信息请参照www.borenpharm.com)(93g,0.29mol),室温过夜,tlc显示反应已完全。反应液加入200ml水,调至酸性,用二氯甲烷萃取,水洗3次,拉干后过柱子得到156.14g黄色油状物,收率88.5%。
[0104]1h nmr(400mhz,cdcl3)δ7.61(d,j=7.3hz,1h),7.53
–
7.42(m,1h),7.41
–
7.07(m,6h),6.23(t,j=5.3hz,1h),5.08(d,j=13.8hz,1h),3.68
–
3.45(m,14h),3.39(dt,j=6.6,5.1hz,2h),3.28(t,j=5.3hz,2h),2.76(ddd,j=16.7,8.3,6.6hz,1h),2.46
–
2.33(m,3h),2.09(dt,j=15.3,6.2hz,1h),1.87(dt,j=16.8,6.2hz,1h),1.38(s,9h).
[0105]
化合物10的合成
[0106]
室温下,将化合物9(35g,57.57mmol)溶于二氯甲烷(150ml)和三氟乙酸(150ml),室温1h,tlc显示反应已完全。反应液拉干过柱子得到24.4g黄色油状物,收率77%。
[0107]
化合物11的合成
[0108]
室温下,将化合物10(40g,72.4mol)缓慢加到化合物n-羟基琥珀酰亚胺(8.58g,74.6mmol)和edci(19.40g,94.1mmol)的二氯甲烷(500ml)溶液中去,室温过夜后加入200ml水,用二氯甲烷萃取,水洗5次,拉干后得到48.7g黄色油状物,收率100%。
[0109]1h nmr(400mhz,cdcl3)δ7.65(d,j=7.4hz,1h),7.56
–
7.43(m,1h),7.43
–
7.11(m,6h),6.30(s,1h),5.13(d,j=13.8hz,1h),3.89
–
3.20(m,19h),2.82(dt,j=23.3,10.6hz,7h),2.54
–
2.33(m,1h),2.14(dt,j=15.1,6.2hz,1h),1.92(dt,j=16.7,6.2hz,1h)。
[0110]
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。