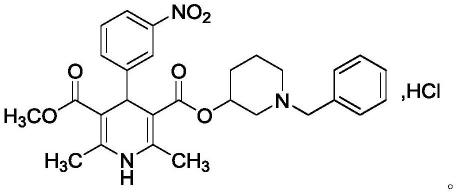
芳基环己基胺衍生物及其制备方法
1.本发明涉及药物合成和药品开发的技术领域。
2.特别地,本发明涉及用于制备环状2-氨基-1-酮衍生物的方法和通过该方法可获得的反应产物和中间体。
3.更特别地,本发明涉及作为所述方法的(中间体)产物的环状3-烯-2-氧基-1-甲酸衍生物和二环氨基甲酸酯衍生物,以及作为最终反应产物和活性药物成分的环状2-氨基-1-酮衍生物,特别地2-氨基环己烷-1-酮衍生物或2-氨基环己烯-1-酮衍生物,它们的互变异构体、立体异构体、盐或溶液。
4.此外,本发明涉及包括环状2-氨基-1-酮衍生物的药物组合物,特别地药物(medicaments)或药品(drugs),以及它们作为药物特别地在预防性或治疗性治疗人或动物身体的疾病,优选地分别神经退行性疾病或精神疾病中的用途。
5.术语神经退行性疾病涵盖特征为中枢神经系统的神经细胞突触或神经元可塑性的逐渐丧失的大多数疾病。神经细胞的这种进行性改变还连续地导致脑的变化,其在神经退行性疾病的进一步进程中本身以各种神经病学和精神病学症状的形式显现。在德国,大约2百万人目前受到神经退行性疾病的影响。由于人口变化,该数量将在2050年增加到超过3百万人。
6.由于脑的神经系统变性过程而发生的一种综合征是痴呆。痴呆被认为是认知、情感和社会能力的恶化加重的各种症状的结合,随着它们的进展,其导致个人的职业受损和后期一般社会功能的受损。自2013年以来,已经使用术语“神经认知疾患”作为术语痴呆的等同或替代。
7.因此,痴呆或神经认知疾患可能由于神经退行性疾病而发生,例如,由于阿尔茨海默病、帕金森病、亨廷顿病或皮克病。此外,痴呆或神经认知疾患还可能由例如颅脑创伤、肿瘤或脑中水保持而引起。
8.痴呆的发展风险随着年龄而增加,由此,最常见的认知疾患形式,称为阿尔茨海默痴呆,通常在六十岁之前不出现。据估计,仅在联邦德国内,大约两百万人目前受痴呆影响。除了老年作为主要的风险因素之外,心血管因素诸如高血压、肥胖症或糖尿病也促进痴呆的发展。痴呆发生的另一个重要因素是抑郁。
9.通常认为抑郁是精神疾病,表达为情绪压抑、持续郁闷和通常缺乏动力感。此外,在日常生活中经常性的观察到减少或丧失了生活乐趣、自尊、表现、共鸣或兴趣。此处决定因素是这些症状不成比例地长时间持续并且与触发症状的因素不成比例。这些触发因素可以是非常多样化的并且可以归因于生物和心理影响两者。德国的抑郁疾患率约8%,其对应于约4百万的成年人。
10.和痴呆一样,抑郁最终是由突触可塑性的丧失所引起,由此突触、神经细胞或者甚至整个脑区域丧失了改变它们的结构和功能以优化进行的过程的能力。还不存在用于治疗特征为神经系统变性过程的突触或神经元可塑性的这种丧失的特定疗法。
11.对于解离的麻醉剂氯胺酮,其是手性芳基环己基胺,观察到它在治疗特别持续性或难以治疗的抑郁上是出乎意料地有效的。静脉内给药氯胺酮的患者在短时间内经历了他
们的抑郁状态的改善,在一些病例中这持续最长达七天。为氯胺酮的抗抑郁效果基础的效果可以最终归因于氯胺酮的代谢物6-羟基去甲氯胺酮(hnk),其中确切的作用方式仍然有争议。然而,已经观察到与氯胺酮相比,6-羟基去甲氯胺酮没有作用于精神的效果,其与氯胺酮相比基本上增加了其作为活性成分使用的可能性。
12.作为在较长时间段内给药例如用于治疗抑郁的药品,因为氯胺酮的解离以及兴奋性和麻醉性效果,因此氯胺酮是不合适的并且携带增加的长期依赖性的风险。此外,氯胺酮包括慢性膀胱毒性并且还可具有肾毒性效应。由于上述原因,氯胺酮仅能够在医院环境中在医疗监督和监视下给药。
13.同样地,尽管其不存在的作用于精神的效果,但是6-羟基去甲氯胺酮不适于用作活性物质或药品。该化合物是高度极性的并且当直接给药时只包括低的生物利用度。在小鼠模型中,例如仅在25mg/kg以及更高的非常高剂量观察到与10mg/kg氯胺酮效果可比较的效果(p zanos et al.,nmdar inhibition-independent antidepressant actions of ketamine metabolites,nature 2016,vol.533(7604),pp.481-6)。6-羟基去甲氯胺酮的高极性使它难以穿过血脑屏障,使得仅不充足地达到作用位点以及因此6-羟基去甲氯胺酮的抗抑郁效果。与氯胺酮一样,可以假定6-羟基去甲氯胺酮的高度的膀胱和肾脏毒性。
14.因此,对于能够用于治疗精神或神经退行性疾病并且具有改善的功效谱的化合物或活性成分,仍然存在未减少的需求。
15.由于氯胺酮或6-羟基去甲氯胺酮的通用功效,已经合成了氯胺酮的衍生物。
16.例如,wo 2019/192602 a1描述了氯胺酮代谢物去甲氯胺酮和6-羟基去甲氯胺酮的不同衍生物以及制备它们的方法。然而,提出的方法仅适用于有限选择的化合物以及特别地不适合于高或多取代的衍生物的合成。
17.此外,所有带有富电子取代基特别地在芳基残基上的化合物,由于它们的不充足的反应性而被排除。所描述的6-羟基去甲氯胺酮衍生物也是高度极性的,其阻止它们作为药品的有效的用途。与母体化合物本身一样,可以预期所描述的6-羟基去甲氯胺酮衍生物具有的生物利用度太低以及过快地从体内排泄。
18.wo 2013/056229 a1还描述了去甲氯胺酮和6-羟基去甲氯胺酮的衍生物以及此外6-羟基氯胺酮的前药。另外,出于此目的而描述的合成方法的适用性或可变性受限,并且例如不适用于空间要求高的取代基或具有富电子基团的取代基。可获得的衍生物的范围因此最终严重受限。
19.此外,极性化合物的前药在实际中常常低效并且它们很难作为药品获得批准。需要额外的合成步骤并且必须证明前药的所有裂解产物是否和如何被代谢。在此上下文中,前药裂解成实际上活性化合物常常导致反应性中间体,这些中间体具有例如毒性特性。
20.因此,在现有技术中描述的氯胺酮和它的代谢物的衍生物在由基本的合成或生产方法可获得的结构方面上严重地受限,其在它们的可变性或灵活性上受限。获得的衍生物通常是高度极性的化合物,这些化合物具有低生物利用度且被快速排泄。因此,这些化合物几乎不适合作为活性剂来治疗例如抑郁。
21.在这一背景下,一种能够制备结构多样的氯胺酮、去甲氯胺酮和6-羟基去甲氯胺酮的衍生物的方法将是高度期望的。此外,因此还特别感兴趣的是,可获得的衍生物提供用于靶向和有效治疗特别涉及中枢神经系统的神经退行性和精神疾患的潜在的新剂。然而,
潜在很好地细胞可渗透的和相应有效的化合物中的很多不能通过至今已知的合成方法来制备,因为特别地芳族化合物上的电子密度表示合成方法的强有力的限制因素。
22.因此,本发明的目的是克服与上文所描述的现有技术相关的问题和缺点,或者至少缓解这些问题和缺点。
23.特别地,本发明的目的是提供一种用于制备氯胺酮或它的代谢物去甲氯胺酮和6-羟基去甲氯胺酮的衍生物的方法。
24.此外,本发明的另外目的是提供氯胺酮或它的代谢物去甲氯胺酮和6-羟基去甲氯胺酮的新的衍生物。
25.根据本发明的第一方面的本发明的主题是一种用于制备根据权利要求1所述的环状2-氨基-1-酮衍生物的方法;本发明的这个方面的另外有利实施方式是各自从属权利要求的主题。
26.根据本发明的第二以及第三方面的本发明的另外主题是根据权利要求10所述的环状3-烯-2-氧基-1-甲酸衍生物,以及根据权利要求11所述的它们的制备方法。
27.另外,根据第四方面的本发明的主题是,根据权利要求12所述的二环氨基甲酸酯,以及,根据根据第五方面的本发明的主题是,根据权利要求13所述的制备它们的方法。
28.此外,根据本发明的第六方面的本发明的主题是,根据权利要求14所述的环状2-氨基-1-酮衍生物。
29.根据本发明的第七以及第八方面的本发明的另外主题是,根据权利要求16所述的环状2-氨基-1-酮衍生物用于刺激或恢复神经元的突触可塑性的用途和根据权利要求17所述的环状2-氨基-1-酮衍生物用于刺激或恢复神经元的神经元可塑性的用途。
30.最后,根据本发明的第九以及第十方面的本发明的主题是根据权利要求18所述的环状2-氨基-1-酮衍生物作为药物的用途以及根据权利要求19所述的包括环状2-氨基-1-酮衍生物的药物组合物的用途。
31.应当理解,下文提到的特定特征,特别地特定实施方式等,其仅是在本发明的一个方面的上下文中描述的,关于本发明的其他方面也是有效的,这不要求明确提及。
32.另外,应当注意关于所有相对的或百分比,特别是重量相关的下文提到的数量数据,在本发明的范围内,这些是本领域技术人员以如此的方式选择使得成分、添加剂或佐剂等的总和结果总是100%或100wt.%。然而,这对于本领域技术人员而言是不言而喻的。
33.此外,它适用于:以下提到的所有参数数据等基本上可以通过标准化或明确规定的确定方法或通过本领域技术人员熟悉的确定方法来确定或查明。
34.在作出的这个附带条件下,下面将更详细地解释本发明的主题。
35.根据本发明的第一方面的本发明的主题是,一种用于制备环状2-氨基-1-酮衍生物的方法,其中在第一反应步骤中将芳族丙烯酸衍生物,特别地碳芳族取代的丙烯酸衍生物,在环化反应中转化为环状3-烯-2-氧基-1-甲酸衍生物。
36.如出人意料地发现,根据本发明的方法提供以特别简单和有效的方式获得大量的不同的环状2-氨基-1-酮衍生物。
37.在本发明的上下文中,衍生物理解为源自一种化学化合物,即所谓的母体化合物,并且因此彼此之间具有密切的结构关系的物质。特别地,衍生物包括与基础母体化合物的官能度或官能团相似的结构单元,其中在衍生物中这些官能度或官能团的至少一个结构元
素呈现的氧化态与母体化合物中的相同。例如,肟和腙是醛和酮的衍生物,或者酯和酰胺是羧酸的衍生物。
38.根据这一定义,作为本发明意义上的母体化合物,可以认为是环状2-氨基-1-酮。相应地,在根据本发明的环状2-氨基-1-酮衍生物中有规律地发现这个结构单元。
39.根据本发明的衍生物的基本结构现在优选源自环己烷,其中也包括包含双键的环状烃,即环己烯。根据本发明的环状2-氨基-1-酮衍生物的基本结构还包括羰基基团和,特别地,靠近羰基基团(即,在根据iupac的2位中)的伯、仲或叔氨基基团。在此上下文中,更优选使用具有伯或叔氨基基团,优选叔氨基基团的衍生物。
40.特别地,因此,根据本发明提供的是,环状2-氨基-1-酮衍生物选自2-氨基环己烷-1-酮衍生物、2-氨基环己烯-1-酮衍生物的组,特别地2-氨基环己烷-1-酮衍生物。
41.根据本发明的生产方法的特定优点特别是,它允许获得大量不同的环状2-氨基-1-酮的衍生物,其中特别地这样的产品还变的易于获得,其不能使用先前的合成方法生产。特别地,带有空间要求高的取代基或富电子取代基的环状2-氨基-1-酮衍生物的制备在先前是不可能的,或者仅在非常有限的范围内可能。特别地,当除了氨基基团之外在2位上引入具有上述提及的取代基的残基时,截止目前尚没有获得将能够以有意义或实际上可靠的方式合成这些衍生物的制备方法。
42.在本发明的上下文中,取代基被理解为原子或原子基团,即,特别地有机残基或官能团,其在分子上替换氢原子,即取代它。特别地,一个或多个氢原子可以被其他原子或原子基团所替换。
43.除了2-氨基基团外引入的残基优选地选自芳族,特别地芳基或杂芳基残基。
44.在本发明的上下文中,芳族、芳族残基或芳族基团特别地被理解为具有连续共轭的电子体系的环状化合物或环状残基或环状基团,其包含4n 2个电子,根据休克尔规则(h
ü
ckel rule)其中n=0或自然数。在此上下文中,芳烃或芳族基团或芳族残基还可以被进一步取代。
45.在本发明的上下文中,术语“芳基”是指它的共轭体系独有地包括碳原子的芳族体系。
46.在本发明的上下文中的术语“杂芳基”优选理解为意指它的共轭体系包括除了碳原子的至少一个原子,即,杂原子的芳族体系。优选地,这种杂原子选自元素周期表中的第3、5和6主族的元素,特别是硼、氮、氧、磷和硫。
47.优选地,因此,环状2-氨基-1-酮衍生物是环状2-氨基-2-芳基-1-酮衍生物或环状2-氨基-2-杂芳基-1-酮衍生物,其中衍生物优选地选自2-氨基-2-芳基-环己烷-1-酮衍生物、2-氨基-2-杂芳基-环己烷-1-酮衍生物、2-氨基-2-芳基-环己烯-1-酮衍生物、2-氨基-2-杂芳基-环己烯-1-酮衍生物的组,特别地2-氨基-2-芳基-环己烷-1-酮衍生物和2-氨基-2-芳基-环己烷-1-酮衍生物。此外,在本发明的上下文中,如果氨基基团选自伯和叔氨基基团,特别地叔氨基基团,获得特别良好的结果。
48.因此,根据本发明的环状2-氨基-1-酮的合适地取代的衍生物还特别地表示为氯胺酮或它的代谢物去甲氯胺酮和6-羟基去甲氯胺酮的衍生物。目前这些化合物只有几种制备方法是可用的,并且就它们的可变性来说它们通常非常有限。
49.现有技术已知的制备方法通常是基于可获得的或已存在的环己酮,然而,其只能
在羰基基团的α位进行有限程度的修饰或官能化。占据空间的基团,诸如取代的芳烃,不能以这种方式结合,因为不利的空间比率阻碍或妨碍相应的反应。因此,现有技术已知的方法就它们的可变性以及因此它们的通用适用性而言严重受限。
50.本发明通过首先建立基本环状结构克服了这种缺点。为了这个目的,进行环化反应,在环化反应中将芳族丙烯酸衍生物转化为环状3-烯-2-氧基-1-甲酸衍生物,即环己烯。在这个反应中,仅芳族丙烯酸衍生物的双键参与,使得特别地在双键的周围,结构变化可以是广泛的。这代表了根据本发明的制备方法的特别和核心优点。
51.因此,在本发明的范围内,首次还可制备这样的环状2-氨基-1-酮衍生物,其带有特别地在结构上要求高或富电子的取代基,使得在根据本发明的方法中具体可变性和灵活性是固有的。
52.特别地,在本发明的范围内,仅用一种生产方法就可获得数量特别大的结构多样的化合物,使得还可以以有利的方式涵盖广范围的具有不同化学、生物或物理特性的化合物。
53.此外,根据本发明的方法特征特别地在于以下的事实,可以从芳族丙烯酸衍生物作为起始点以仅少许方法或反应步骤获得环状2-氨基-1-酮衍生物。此外,相对少的反应步骤优选不需要任何复杂的清洁步骤,使得根据本发明的制备方法还是很省时和易于进行的。另外,在该方法中使用的起始化合物以及试剂和溶剂可以特别地以不复杂的方式获得或者可以容易地合成制备。
54.此外,单独反应步骤包括特别地高度的特异性和选择性,使得根据本发明的生产方法可以以针对性的方式进行并且没有显著的副产物的产生。因此,根据本发明的制备方法还是特别地用户友好和易于操作的。
55.在根据本发明的方法中获得的中间体的进一步特征特别地在于以下事实:它们允许大范围的额外的修饰或官能化。例如,在第一反应步骤中获得的环状3-烯-2-氧基-1-甲酸衍生物可以通过修饰环己烯骨架的双键而进行修饰。这特别允许直接且不复杂地获得环状2-氨基-1-酮衍生物,或者它们的前体,其包括不同的取代模式。特别地,使用用于目标化合物类别,即根据本发明的环状2-氨基-1-酮衍生物的根据本发明的方法,首次使得这样的合成灵活性以及允许这样程度的结构多样性成为可能。
56.因此,根据本发明的制备方法还使能够特别地关于环状2-氨基-1-酮衍生物(提供大量不同的潜在新活性成分)例如用于治疗神经退行性疾病,特别地痴呆,或精神疾病,特别地抑郁的用途。特别地,还有利的是,本发明的环状2-氨基-1-酮衍生物可以包括更平衡的亲水性和亲脂性分布图,使得促进它们摄取进入身体,以及特别地促进它们穿过血脑屏障。
57.在本发明的范围内,因此首次可以生产广范围的氯胺酮、去甲氯胺酮和羟基去甲氯胺酮衍生物以及其他结构相似的化合物。特别地,可以容易地调节目标化合物的极性,并且能够获得包括比氯胺酮、去甲氯胺酮或羟基去甲氯胺酮显著地更低极性的衍生物。一方面,这导致了潜在较高的功效,特别地关于神经退行性疾病的治疗,因为显著地增加了根据本发明的衍生物的生物利用度,特别地它们的穿过细胞膜或血脑屏障的能力。另一方面,本发明的化合物在体内的停留时间也可以增加,其特别地由于衍生物的更慢的代谢或肾排泄。原则上,这允许获得包括改善的功效和更少副作用,诸如减少的膀胱毒性的新活性成分
和药品。
58.关于根据本发明的方法的第一反应步骤,现在优选地提供的是环化反应是环加成,特别地[4 2]环加成,优选地第尔斯-阿尔德(diels-alder)反应。
[0059]
在本发明的上下文中,环化反应通常理解为产生环状产物的反应。该反应可以从一种或多种起始化合物开始,其在环化反应中反应,例如,构成四元环、五元环、六元环等。根据本发明,更优选的是,如果环化反应以双分子方式进行,即两个分子参与。
[0060]
另外,在本发明的上下文中,[4 2]环加成理解为这样类型的反应,其中起始化合物的四个电子和第二起始化合物的两个电子参与反应。[4 2]环加成的特定情况是第尔斯-阿尔德反应,在该反应中通常二烯,即具有四个电子的起始化合物,和亲双烯体,即具有两个电子的起始化合物,相互反应以形成环己烯衍生物。
[0061]
在这种意义上,芳族丙烯酸衍生物优选是根据本发明的亲双烯体。因此,根据描述的用于环化反应的优选的实施方式,特别地提供的是,通过与二烯反应将芳族丙烯酸衍生物转化为环状3-烯-2-氧基-1-甲酸衍生物。
[0062]
在这方面,在本发明的上下文中,如果通过与1,3-丁二烯醇衍生物反应将芳族丙烯酸衍生物转化为环状3-烯-2-氧基-1-甲酸衍生物,获得特别良好的结果。因此,根据本发明的这个实施方式,优选使用1,3-丁二烯醇衍生物作为环化反应中的二烯。
[0063]
根据本发明,进一步优选的是如果芳族丙烯酸衍生物选自通式i的化合物
[0064][0065]
其中
[0066]
r1=芳基、杂芳基;
[0067]
pg=保护基团。
[0068]
关于残基r1,如果,对于r1=芳基,r1选自环状芳基残基,特别地选自萘基、蒽基、菲基残基和通式ii的芳基残基的组,特别地已经证明是很好的
[0069][0070]
其中,各自彼此独立地,
[0071]r11
、r
12
、r
13
=h;
[0072]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、f、cl;
[0073]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0074]
cooh、-conh2、cosh、cho,特别地cooh、conh2;
[0075]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0076]
苯基、萘基;
[0077]
其中
[0078]
x=2至20,特别地2至15,优选地2至10。
[0079]
优选地,对于r1=芳基,r1选自通式ii的芳基残基。此外,在本发明的范围内,特别地关于残基r
11
、r
12
、r
13
,更优选的是如果x=2至5,特别地2至3。
[0080]
同样地,关于残基r1的结构或组成,可以提供的是,对于r1=杂芳基,r1选自以下的组:5-环杂芳基,特别地吡咯基、咪唑基、吡唑基、噁唑基、异噁唑基、噻唑基、呋喃基、噻吩基残基和其取代的衍生物,优选吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物,和/或6元杂芳基,特别地吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物,优选吡啶基残基及其取代的衍生物。
[0081]
在本发明的上下文中,特别地优选如果r1=杂芳基选自噻吩基残基、吡啶基残基及其取代的衍生物的组。
[0082]
在本发明的上下文中,进一步优选的是,杂芳基具有选自以下的组的取代基:烷基残基,特别地,甲基、乙基、丙基残基,卤烷基残基,特别地氟烷基、氯烷基残基,全氟烷基残基,特别地三氟甲基、五氟乙基残基,烷氧基残基,特别地甲氧基、乙氧基、丙氧基残基,全氟烷氧基残基,特别地三氟甲氧基、五氟乙氧基残基,和/或硝基残基。这适用于本发明的其中r1=杂芳基的所有实施方式。
[0083]
特别地,本发明因此允许大量的用于根据本发明的方法中的具有各种各样的取代模式或不同杂原子的芳族结构。以这种方式,特别地,获得用于根据本发明的方法的特别地高的丰富度的变体和高的灵活性。
[0084]
根据本发明的方法的这种特别优点是特别地基于以下事实:使能够简单衍生化和合成各种活性成分的环状2-氨基-1-酮衍生物的己烷或己烯主链没有建立,直到根据本发明的方法,特别地环化反应。有利地,仅仅相关的双键参与环化反应。特别地,这开辟了残基r1的宽范围的选择,因为它对于反应过程几乎没有影响并且因此可以被构型为,例如,占据空间的或推电子的。
[0085]
关于芳族丙烯酸衍生物的羧酸官能团,根据本发明优选提供的是,这用保护基团(pg)封端,优选用后者进行酯化。
[0086]
在本发明的上下文中,将保护基团(pg)理解为意指特别地在化学合成期间,特别地多阶段化学合成期间被引入到化合物中的特定官能团上的取代基,以便暂时保护特定官能团和因此阻止在该基团上的不希望的反应。
[0087]
如上文所提及,为了这个目的优选将羧酸酯化或特别地转化成酯。关于用于这个目的的保护基团(pg),它通常可选自大量的不同保护基团或取代基,特别地其中该选择基于各自预期的反应条件。
[0088]
特别地,保护基团(pg)选自有机残基。有机残基被理解为主要由碳、氢和氧,以及任选地氮、硫、磷等构成的取代基或基团。此外,有机残基可以包括官能团。例如,根据本发明,可以提供的是,用包括羧酸或羧酸衍生物的有机残基作为官能团封端或酯化芳族丙烯酸衍生物的羧酸官能团,特别地,其中产生碳酸酐。更优选地,然而,在本发明的范围内,芳族丙烯酸衍生物的羧酸官能团被转化为酯。
[0089]
合适的保护基团或取代基可以包括,例如,烷基残基、芳基残基或官能单元诸如辅基(auxiliaries)或可比的占据空间的或空间要求高的基团。
[0090]
在本发明的上下文中,辅基被理解为适于影响随后反应的立体化学过程的取代基或基团。特别地,手性辅基可以以这样的方式影响本身非立体选择性或不是非常立体选择性的反应的过程,使得无论在反应侧是没有给予或几乎没有给予的选择性,与用手性辅基保护的化合物相关而过量或理想地唯一形成非对映异构体,或与裂解辅基之后获得的化合物相关,形成期望的手性化合物的对映异构体。
[0091]
适合作为根据本发明的保护基团(pg)的辅基可以选自,例如,evans辅基、薄荷基辅基、enders试剂、衍生字这些的衍生物及其混合物的组。
[0092]
此外,如果保护基团(pg)包括芳基残基,即芳族基团,那么获得了特别地还与根据本发明的方法中的选择性或特别地立体选择性相关的特别良好的结果。
[0093]
此外,更优选保护基团(pg)特别地包括在相对温和条件下可以裂解的保护基团或特别地酯基团。
[0094]
如果保护基团(pg)选自ph不稳定的保护基团和/或氧化还原不稳定的保护基团的组,特别地选自苄基-、对-甲氧基苄基-、二甲氧基苄基、烷氧基羰基、三苯基甲基、烷基、烯丙基基团的组,优选对-甲氧基苄基、二甲氧基苄基、烷氧基羰基、三苯基甲基基团,更优选对-甲氧基苄基、烷氧基羰基基团,已经证明是很好的。
[0095]
与对-甲氧基苄基和烷氧基羰基基团均等或除对-甲氧基苄基和烷氧基羰基基团之外,更优选保护基团选自烷基基团,特别地选自甲基、乙基、丙基基团的组,优选甲基基团。
[0096]
上文提及的保护基团的特别地特征在于以下事实:它们可以以不复杂的方式且高产率以及可靠地限制羧酸官能团的反应性来引入。同时,上文提及的保护基团可以在相对温和的条件下几乎完全去除,其中相应的条件可以特别好地整合进根据本发明的生产方法中。
[0097]
对于根据本发明的制备方法,如果芳族丙烯酸衍生物选自通式iii的化合物,现在已经证明是有利的
[0098][0099]
其中
[0100]
r1=芳基、杂芳基;
[0101]
其中,对于r1=芳基,r1选自通式iv的芳基残基
[0102][0103]
其中,各自彼此独立地,
[0104]r11
、r
12
、r
13
=h;
[0105]
nh2、oh、f、cl;
[0106]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0107]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0108]
以及
[0109]
x=2至10,特别地2至5,优选地2至3;
[0110]
以及其中对于r1=杂芳基,r1选自吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物和/或吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物的组;
[0111]
pg=ph不稳定的保护基团和/或氧化还原不稳定的保护基团,特别地对-甲氧基苄基、二甲氧基苄基、烷氧基羰基、三苯基甲基基团。
[0112]
在本发明的非常特别优选的实施方式中,进一步优选的是如果芳族丙烯酸衍生物选自通式v的化合物
[0113][0114]
其中
[0115]
r1=芳基、杂芳基;
[0116]
其中,对于r1=芳基,r1选自通式vi的芳基残基
[0117][0118]
其中,各自彼此独立地,
[0119]r11
、r
12
、r
13
=h;
[0120]
nh2、oh、f、cl;
[0121]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0122]
以及
[0123]
x=2至5,特别地2至3;
[0124]
以及其中对于r1=杂芳基,r1选自噻吩基残基、吡啶基残基以及其取代的衍生物的组;
[0125]
pg=ph不稳定的保护基团和/或氧化还原不稳定的保护基团,特别地对-甲氧基苄基、二甲氧基苄基、烷氧基羰基。
[0126]
只要涉及1,3-丁二烯醇衍生物,在本发明的上下文中,这通常可以选自技术人员本身已知的1,3-丁二烯醇的衍生物。根据本发明,然而,如果1,3-丁二烯醇衍生物选自富电子1,3-丁二烯醇衍生物,特别地选自通式vii的1,3-丁二烯醇衍生物,已经证明是很好的
[0127][0128]
其中
[0129]r‘
=c
1-c
x-烷基、c
1-c
x-乙烯基、c
1-c
x-烯丙基;
[0130]
苯基、苄基;
[0131]
三烷基硅烷基;
[0132]
以及
[0133]
x=2至10。
[0134]
特别优选地,在本发明的上下文中,1,3-丁二烯醇衍生物选自通式viii的丁二烯氧基硅烷
[0135][0136]
其中
[0137]r‘
=三烷基硅烷基;
[0138]
三烷基硅烷基,特别地三甲基硅烷基、三乙基硅烷基、三异丙基硅烷基、叔丁基二甲基硅烷基、三苯基硅烷基、叔丁基二苯基硅烷基;优选三乙基硅烷基、三异丙基硅烷基、叔丁基二甲基硅烷基。
[0139]
在本发明的上下文中,富电子1,3-丁二烯醇衍生物的使用特别地确保二烯的反应性足够高使得环化反应优选地快速和完全进行。此外,可以以有利的方式补偿亲双烯体(即芳族丙烯酸衍生物)的可能过高的电子密度或电子态的差异可以特别地用于有利于反应的充分有利的过程。特别地,上文提及的残基r'有助于以下事实:增加二烯中的电子密度。
[0140]
关于反应条件,在这些反应条件下进行环化反应,或特别地进行芳族丙烯酸衍生物与1,3-丁二烯醇衍生物的反应,这些可以在本发明的上下文中选自技术人员本身已知的用于环化的反应条件。
[0141]
根据本发明,如果芳族丙烯酸衍生物与1,3-丁二烯醇衍生物的反应是以在0.5:1至1:20、特别地1:1至1:15、优选地1:2至1:10的范围内的芳族丙烯酸衍生物与1,3-丁二烯醇衍生物的比率进行,特别地获得良好的结果。
[0142]
此外,如果反应是在惰性溶剂中进行的,特别地环状的,特别地芳族的、烃,优选四氢呋喃、二甲苯或其混合物的溶剂中进行的,已经证明是有用的。
[0143]
根据本发明,惰性溶剂理解为本身不参与反应的溶剂。但是,可能的是该溶剂在一定程度上影响反应的过程,例如通过以某种,特别地有利的方式使起始化合物配位,例如通过可能的相互作用,以这样的方式使得有利于或特别地加速环化。
[0144]
此外,在本发明的范围内,如果反应在供热下进行,特别地在50至250℃、优选75至200℃、更优选100至175℃的范围内的温度下进行,已经证明是很好的。
[0145]
在上文提及的温度范围内,特别地观察到根据本发明的制备方法的高的或者完全的转化,以及环化反应的同样高的选择性和特异性。
[0146]
此外,根据本发明优选的是如果反应在5至300min,特别地10至250min,优选地15至200min的时间段内进行。
[0147]
在上文提及的反应持续时间的范围内,通常可以观察到起始化合物完全转化为环化产物,然而产生的副产物可忽略不计。
[0148]
最终,在本发明的上下文中,如果反应是在自由基清除剂的存在下,特别地在二羟基苯的存在下,优选地在氢醌的存在下进行,已经发现是特别有利的。
[0149]
在本发明的上下文中,特别地关于自由基清除剂添加剂,已经观察到可以以有利的方式改善起始化合物彼此之间的反应,在于干预副产物的产生,特别地有效地抑制特别
地聚合的副产物。
[0150]
对于第一反应步骤,根据本发明现在特别地提供的是,获得的环状3-烯-2-氧基-1-甲酸衍生物选自通式ix的化合物
[0151][0152]
其中
[0153]
r1=芳基、杂芳基;
[0154]
特别地,其中对于r1=芳基,r1选自环状芳基残基,特别地选自萘基、蒽基、菲基残基和通式ii的芳基残基的组
[0155][0156]
其中,各自彼此独立地,
[0157]r11
、r
12
、r
13
=h;
[0158]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、f、cl;
[0159]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0160]
cooh、-conh2、cosh、cho,特别地cooh、conh2;
[0161]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0162]
苯基、萘基;
[0163]
其中
[0164]
x=2至20,特别地2至15,优选地2至10;
[0165]
以及,特别地,其中对于r1=杂芳基,r1选自以下的组:5元环杂芳基,特别地吡咯基、咪唑基、吡唑基、噁唑基、异噁唑基、噻唑基、呋喃基、噻吩基残基和其取代的衍生物,优选吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物,和/或6元杂芳基,特别地吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物,优选吡啶基残基及其取代的衍生物;
[0166]
pg=ph不稳定的保护基团和/或氧化还原不稳定的保护基团,特别地苄基、对-甲氧基苄基、二甲氧基苄基、烷氧基羰基、三苯基甲基、烷基、烯丙基基团,更优选对-甲氧基苄基、二甲氧基苄基、烷氧基羰基、三苯基甲基基团,更优选对-甲氧基苄基、烷氧基羰基基团;
[0167]r‘
=三烷基硅烷基,特别地三甲基硅烷基、三乙基硅烷基、三异丙基硅烷基、叔丁基二甲基硅烷基、三苯基硅烷基、叔丁基二苯基硅烷基;优选三乙基硅烷基、三异丙基硅烷基、叔丁基二甲基硅烷基。
[0168]
同样地,在本发明的上下文中,如果环状3-烯-2-氧基-1-甲酸衍生物选自通式x的化合物,已经证明是很好的
[0169][0170]
其中
[0171]
r1=芳基、杂芳基
[0172]
其中,对于r1=芳基,r1选自通式iv的芳基残基
[0173][0174]
其中,各自彼此独立地,
[0175]r11
、r
12
、r
13
=h;
[0176]
nh2、oh、f、cl;
[0177]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0178]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0179]
以及
[0180]
x=2至10,特别地2至5,优选地2至3;
[0181]
以及其中对于r1=杂芳基,r1选自吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物和/或吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物的组;
[0182]
pg=ph不稳定的保护基团和/或氧化还原不稳定的保护基团,特别地对-甲氧基苄基、二甲氧基苄基、烷氧基羰基、三苯基甲基基团;
[0183]r‘
=三乙基硅烷基、三异丙基硅烷基、叔丁基二甲基硅烷基。
[0184]
在根据本发明的方法的更优选的实施方式中,进一步提供的是,环状3-烯-2-氧基-1-甲酸衍生物选自通式xi的化合物
[0185][0186]
其中
[0187]
r1=芳基、杂芳基
[0188]
其中,对于r1=芳基,r1选自通式vi的芳基残基
[0189][0190]
其中,各自彼此独立地,
[0191]r11
、r
12
、r
13
=h;
[0192]
nh2、oh、f、cl;
[0193]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0194]
以及
[0195]
x=2至5,特别地2至3;
[0196]
以及其中对于r1=杂芳基,r1选自噻吩基残基、吡啶基残基以及其取代的衍生物的组;
[0197]
pg=ph不稳定的保护基团和/或氧化还原不稳定的保护基团,特别地对-甲氧基苄基、二甲氧基苄基、烷氧基羰基;
[0198]r‘
=三乙基硅烷基、三异丙基硅烷基、叔丁基二甲基硅烷基。
[0199]
在根据本发明的方法的另外过程中,现在优选提供的是,在第一反应步骤中的环状3-烯-2-氧基-1-甲酸衍生物的生产之后是第二反应步骤。在此上下文中,如果在第二反应步骤中将环状3-烯-2-氧基-1-甲酸衍生物转化为二环氨基甲酸酯衍生物,已经证明是很好的。
[0200]
在此上下文中,可以优选提供的是,在第二反应步骤的子步骤中,将环己烯骨架,特别地环己烯骨架的双键优选地官能化为化合物xii
[0201][0202]
其中残基r4、r5、r6,特别地其中所述残基r4、r5、r6各自彼此独立地,选自以下的组:
[0203]
r4、r5、r6=h;
[0204]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、sh;
[0205]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0206]
cooh、conh2、cosh、cho,特别地cooh、conh2;
[0207]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0208]
其中
[0209]
x=2至20,特别地2至15,优选地2至10,更优选地2至5,更优选地2至3;以及
[0210]
y=0、1,其中对于残基r4、r5、r6中的至少一个,y=1。
[0211]
因此,在根据本发明的方法的骨架内,可以在制备方法的早期阶段进行环己烷主链的进一步修饰或官能化,使得以有利的方式在环状3-烯-2-氧基-1-甲酸衍生物的阶段处仍然可获得大量结构不同的化合物。
[0212]
如果通过根据本发明的方法提供具有不同的取代模式的环状2-氨基-1-酮衍生物,因为这种形式的官能化或修饰通过双键的目标反应特别容易地且直接地可获得,这是特别有利的。
[0213]
此外,根据本发明如果在第二反应步骤的另外子步骤中,羧酸基团通过叠氮化转化为羧酸叠氮化物,已经证明是很好的。
[0214]
在本发明的上下文中,叠氮化理解为意指叠氮化物基团的引入,特别地在羧酸官能团处。因此,羧酸叠氮化物是羧酸的衍生物并且包括通用结构r-c(o)n3。
[0215]
根据本发明,如果在叠氮化之前的反应步骤中,优选地在还原和/或碱性或酸性条
件下去除保护基团(pg)和/或残基r’,特别地保护基团(pg)和残基r’,已经证明是有利的。
[0216]
在这种意义上,根据本发明更优选的是,如果保护基团(pg)和/或残基r’,特别地保护基团(pg)和残基r’的去除,氢解地,即通过在金属催化剂上在氢气的存在下还原,和/或在碱性或酸性条件下,优选地在氟离子的存在下,例如使用四丁基氟化铵(tbaf)进行。同样地,然而,在本发明的范围内,还可以使用本领域技术人员的常用方案用于保护基团的去除。
[0217]
对于第二反应步骤,在本发明的上下文中,如果使用选自以下的组的试剂进行叠氮化:无机叠氮化物,特别地叠氮化钠,和/或有机叠氮化物,特别地磺酰叠氮、磷酰叠氮,优选地对甲苯磺酰叠氮、二苯基磷酰叠氮,或其混合物,还已经证明是很好的。
[0218]
特别地,在根据本发明的方法中,如果用于叠氮化的试剂是以过量1至20倍,特别地过量1至10倍,优选地过量1.5至7倍使用,在每种情况下均基于所使用的环状3-烯-2-氧基-1-甲酸衍生物的量,获得良好的结果。
[0219]
使用的叠氮化物允许特别地羧酸官能团的温和叠氮化以及还具有可以容易地和廉价地分离由试剂形成的副产物的优点。因此,在第二反应步骤的范围内,可以有利地摒弃复杂的清洁或分离程序。
[0220]
此外,对于根据本发明的方法,如果在碱,特别地有机碱,优选地氮碱,更优选地叔胺,更优选地三乙胺的存在下进行叠氮化,获得特别良好的结果。
[0221]
替代地,在本发明的范围内可以使用其他碱,特别地氮碱,其中合适的碱,特别地氮碱本身为本领域技术人员熟悉。在本发明的上下文中特别地优选的通常是,那些仅包括低亲核特性的碱,如例如对于更高取代的胺的情况。
[0222]
结合上文提及的叠氮化物试剂,因此可以特别地在本发明的范围内提供叠氮化的温和条件,其特别地还能够很好地耐受如之前列举的另外可能的取代基。同时,上文提及的试剂的使用导致特别地高的转化率以及羧酸的极度特定或选择的反应性。
[0223]
只要涉及溶剂的选择,这通常可以从本领域技术人员通常使用的溶剂中进行选择。特别地,唯一相关的是,溶剂能够充分溶解起始材料和用于叠氮化的试剂以及同时不参与叠氮化。
[0224]
在本发明的上下文中,如果叠氮化是在惰性溶剂中进行,特别地由环状,特别地芳族烃,优选地甲苯组成的溶剂中进行,获得特别良好的结果。
[0225]
此外,对于根据本发明的方法,优选的是如果叠氮化在供热下进行,特别地在40至200℃、优选50至150℃、更优选60至125℃的范围内的温度下进行。
[0226]
此外,如果叠氮化在1小时至48小时,特别地1.5至36小时,优选地1.5至24小时的时间段内进行,已经证明是很好的。
[0227]
特别地,在本发明的上下文中,在上文提及的温度或时间范围内可以观察到起始化合物的非常良好或完全的转化,没有记录到任何副产物的形成。在这种意义上,第二反应步骤还优选地特征在于高选择性和特异性,并且对于宽选择的不同起始化合物,还相应地可以可靠地进行。
[0228]
最终,对于转化为二环氨基甲酸酯衍生物,如果在第二反应步骤的另外子步骤中,羧酸叠氮化物环化为氨基甲酸酯,特别地通过重排环化为对应的异氰酸酯,已经证明是特别有利的。
[0229]
根据本发明,氨基甲酸酯产生特别地发生在原位,即作为叠氮化的结果直接成为羧酸叠氮化物。环化优选地基于先前的羧酸叠氮化物根据所称的库尔修斯(curtius)反应重排成为对应的异氰酸酯。然后,在二环氨基甲酸酯衍生物之后的涉及相邻羟基基团的分子内反应,以针对性的方式以及特别地以高产率产生对应的二环氨基甲酸酯衍生物。
[0230]
根据本发明的方法的特别优点在于,取决于起始材料的构型,氨基甲酸酯的环化,其可以是例如内型-和/或外型-构型,产生顺式-/反式-(syn-/anti-)异构体混合物,但是由于这一点,通常可以摒弃异构体的耗时的分离。在根据本发明的方法的另外过程中,通常可以进一步等效地使用所有异构体,因为通常随后将1-羟基基团氧化为酮。
[0231]
在此上下文中,根据本发明更优选地提供的是,二环氨基甲酸酯衍生物选自通式xiii的衍生物
[0232][0233]
其中
[0234]
r1=芳基、杂芳基;
[0235]
特别地,其中对于r1=芳基,r1选自环状芳基残基,特别地选自萘基、蒽基、菲基残基和通式ii的芳基残基的组
[0236][0237]
其中,各自彼此独立地,
[0238]r11
、r
12
、r
13
=h;
[0239]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、f、cl;
[0240]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0241]
cooh、-conh2、cosh、cho,特别地cooh、conh2;
[0242]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0243]
苯基、萘基;
[0244]
以及,其中对于r1=杂芳基,r1选自以下的组:5元环杂芳基,特别地吡咯基、咪唑基、吡唑基、噁唑基、异噁唑基、噻唑基、呋喃基、噻吩基残基和其取代的衍生物,优选吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物,和/或6元杂芳基,特别地吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物,优选吡啶基残基及其取代的衍生物;
[0245]
r2、r3=各自彼此独立地,
[0246]
h;
[0247]c1-c
x-烷基;c
1-c
x-环烷基;
[0248]
r4、r5、r6=各自彼此独立地,
[0249]
h;
[0250]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、sh;
[0251]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0252]
cooh、conh2、cosh、cho,特别地cooh、conh2;
[0253]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0254]
其中
[0255]
x=2至20,特别地2至15,优选地2至10;以及
[0256]
y=0、1。
[0257]
此外,在本发明的上下文中,如果二环氨基甲酸酯衍生物选自通式xiv的衍生物,已经证明是有利的
[0258][0259]
其中
[0260]
r1=芳基、杂芳基;
[0261]
其中,对于r1=芳基,r1选自通式iv的芳基残基
[0262][0263]
其中,各自彼此独立地,
[0264]r11
、r
12
、r
13
=h;
[0265]
nh2、oh、f、cl;
[0266]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0267]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0268]
以及其中对于r1=杂芳基,r1选自吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物和/或吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物的组;
[0269]
r4、r5、r6=各自彼此独立地,
[0270]
h;
[0271]
nh2、oh、sh;
[0272]c1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0273]
其中
[0274]
x=2至10,特别地2至5,优选地2至3;以及
[0275]
y=0、1。
[0276]
此外,根据本发明的更优选的实施方式,可以提供的是,二环氨基甲酸酯衍生物选自通式xv的衍生物
[0277][0278]
其中
[0279]
r1=芳基、杂芳基;
[0280]
其中,对于r1=芳基,r1选自通式vi的芳基残基
[0281][0282]
其中,各自彼此独立地,
[0283]r11
、r
12
、r
13
=h;
[0284]
nh2、oh、f、cl;
[0285]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0286]
以及其中对于r1=杂芳基,r1选自噻吩基残基、吡啶基残基以及其取代的衍生物的组;
[0287]
r4、r5、r6=各自彼此独立地,
[0288]
h;
[0289]
nh2、oh、sh;
[0290]c1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-全氟烷氧基;
[0291]
其中
[0292]
x=2至5,特别地2至3;以及
[0293]
y=0、1。
[0294]
在本发明的上下文中,特别地关于残基r4、r5、r6,因此可以优选地提供的是,二环氨基甲酸酯包括环己烯骨架或环己烷骨架。在二环氨基甲酸酯包括环己烯骨架的情况下,y=0特别地适用于残基r4、r5。如果二环氨基甲酸酯包括环己烷骨架,y=1特别地适用于残基r4、r5。这在上文提及的结构式中由到残基r4、r5的虚线键或虚线双键示出。
[0295]
从在第二反应步骤中获得的二环氨基甲酸酯衍生物开始,在本发明的上下文中现在优选提供的是,在第三反应步骤中将二环氨基甲酸酯衍生物转化为环状2-氨基-1-酮衍生物,特别地2-氨基环己烷-1-酮衍生物或2-氨基环己烯-1-酮衍生物,优选地2-氨基环己烷-1-酮衍生物。
[0296]
为了这个目的,如果在第三反应步骤的子步骤中,在单环2-氨基-1-醇衍生物的形成下去除氨基甲酸酯基团,首先已经证明是很好的。
[0297]
根据本发明,如果在还原和/或碱性条件下,特别地在碱金属盐,优选地钠盐、锂盐或其混合物,优选地锂盐的存在下,进行氨基甲酸酯基团的去除,在此获得特别良好的结
果。
[0298]
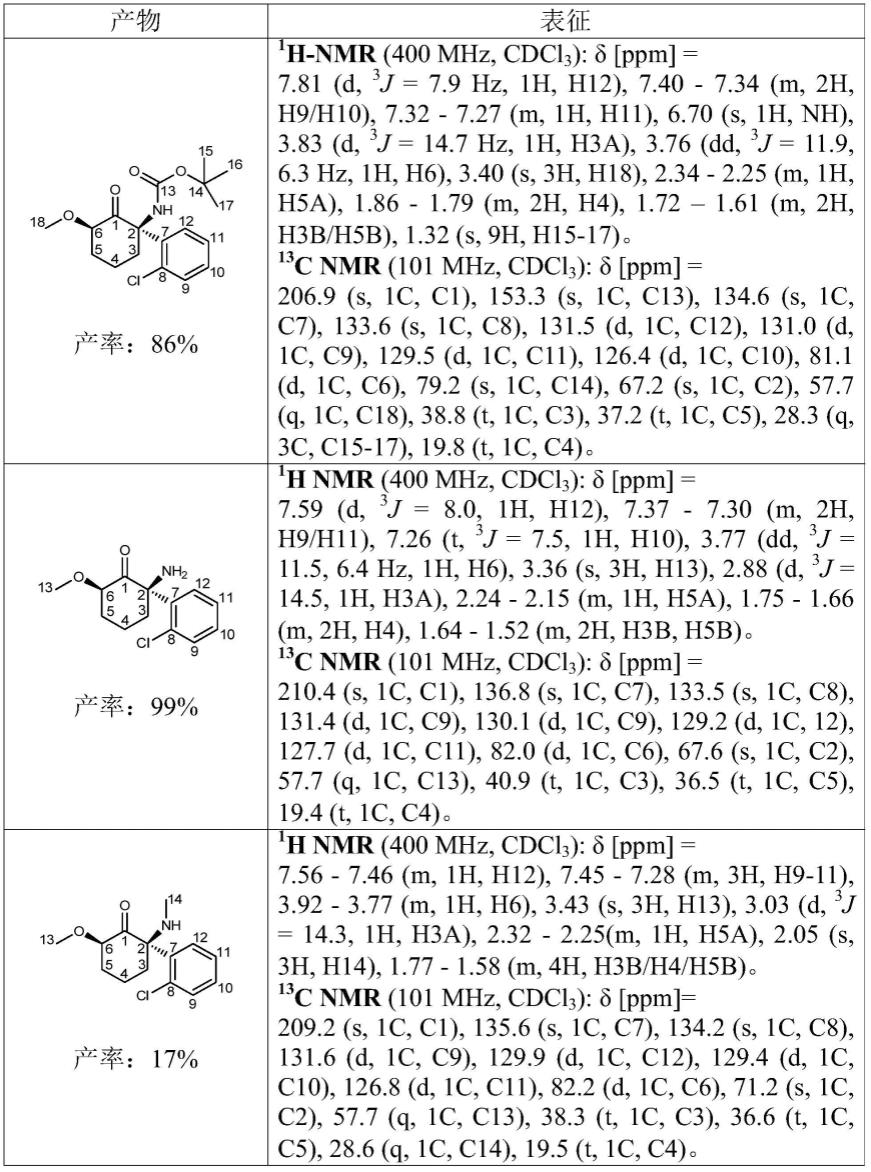
特别地,更优选地使用的是,如果碱金属盐,特别地锂盐,选自氢氧化物和/或四氢合金属酸盐(tetrahydridometallates)的组,特别地氢氧化锂和/或氢化铝锂。
[0299]
在此上下文中,如果在极性溶剂中,特别地环状的,特别地含氧烃,优选地四氢呋喃、二噁烷或它们的混合物的溶剂中进行氨基甲酸酯基团的去除,已经证明是有利的。
[0300]
同样地,如果在在-10至250℃、优选-5至200℃、更优选0至175℃的范围内的温度下进行氨基甲酸酯基团的去除,已经证明是很好的。
[0301]
此外,优选地提供的是,反应在10分钟至48小时,特别地15分钟至36小时,优选地20分钟至24小时的时间段内进行。
[0302]
在上文提及的反应条件下,特别地在上文提及的时间或反应持续时间内,可以实现氨基甲酸酯基团的可靠且完全的去除,没有观察到任何实质上的副产物的产生。
[0303]
同样地,根据本发明的方法的特别的优点是,取决于还原和/或碱性条件,特别地碱金属盐的选择,可以获得不同的反应产物,诸如,例如,伯胺或甲基取代的仲胺。因此,根据本发明的方法允许特别地简单和直接地获得氯胺酮或其代谢物去甲氯胺酮和6-羟基去甲氯胺酮的衍生物。因此还可以将讨论中的反应步骤理解为,特别地以某种方式作为分化点,使得可以易于获得大量的不同的氯胺酮的代谢阶段。因此,与碱,诸如特别地氢氧化锂的反应,选择性地导致伯胺的产生,然而还原条件,诸如特别地与氢化铝锂(lialh4)反应,选择性地产生仲胺作为产物。
[0304]
在这个子步骤之后,根据本发明现在优选地提供的是,在第三反应步骤的另外子步骤中,氧化羟基基团,产生2-氨基-1-酮衍生物。
[0305]
在此上下文中,如果在酸性条件下,特别地在铬(vi)化合物的存在下进行氧化,已经证明是有利的。
[0306]
在这一点上,根据本发明特别地优选的是,如果铬(vi)化合物是氧化铬(vi),特别地溶解在硫酸中。优选地,在2-氨基-1-酮衍生物的形成下,即,在对应于琼斯(jones)氧化的那些条件的条件下,进行羟基基团的氧化。在此上下文中,特别出人意料地发现,琼斯氧化尤其适于实现期望的羟基基团氧化为酮,但是,替代的氧化主要产生相应的特别地不希望的n-氧化物。
[0307]
为了这个目的,进一步优选的是,如果在极性溶剂中,特别地低沸点溶剂中,优选地二氯甲烷、丙酮或其混合物中进行氧化。
[0308]
另外,如果在-30至60℃、优选-25至50℃、更优选-20至45℃的范围内的温度下进行氧化,已经证明是很好的。
[0309]
最后,在本发明的上下文中,如果反应在5分钟至24小时,特别地10分钟至18小时,优选地15分钟至12小时的时间段内进行,获得特别良好的结果。
[0310]
如果期望环状2-氨基-1-酮衍生物的进一步多样化,即修饰或官能化,在本发明的上下文中在第三反应步骤的另外子步骤中将2-氨基基团官能化,特别地烷基化,优选地双烷基化也可以是适当的。
[0311]
在这种情况下,如果使用选自c
1-c
x-烷基、c
1-c
x-环烷基,特别地其中x=2至20,优选地2至15,更优选地2至10,进一步优选地2至5,更优选地2至3的组的残基进行官能化,特别地烷基化,优选地双烷基化,特别地已经证明是很好的。在此上下文中,更优选地是,特别
地对于双烷基化,如果残基各自彼此独立地选择。
[0312]
在本发明的更优选的实施方式的上下文中,还可能的是,特别地对于双烷基化,一起选择残基。然后,优选地提供的是,残基选自以下的组:c
1-c
x-环烷基,其中x=2至10,特别地2至5,优选地2至3,优选地吡咯烷基、哌啶基、咪唑基、吡啶基残基及它们的取代的衍生物。
[0313]
同样地,在本发明的更优选的实施方式的骨架内,如果没有已经在根据本发明的制备方法的早期阶段中进行,可以在第三反应步骤的另外子步骤中,将环己烯骨架,特别地环己烯骨架的双键优选地官能化为化合物xvi
[0314][0315]
其中残基r4、r5、r6,特别地其中残基r4、r5、r6各自彼此独立地,选自以下组:
[0316]
r4、r5、r6=h;
[0317]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、sh;
[0318]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0319]
cooh、conh2、cosh、cho,特别地cooh、conh2;
[0320]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0321]
其中
[0322]
x=2至20,特别地2至15,优选地2至10,更优选地2至5,更优选地2至3;以及
[0323]
y=0、1,其中对于残基r4、r5、r6中的至少一个,y=1。
[0324]
关于环己烯骨架的进一步官能化,一方面在本发明的上下文中特别地优选的是,如果将环己烯的骨架的双键官能化,特别地获得对应的环己烷衍生物。
[0325]
已经证明本发明的特别优点是,在根据本发明的方法的骨架内可以获得或使用具有环己烯骨架的化合物,特别地其中已经示出这些化合物可以特别地通用的方式修饰和官能化。在这个基础上,可以从例如,仅一种环己烯化合物获得大量的非常不同的环己烷衍生物,以及相应地,可以提供大范围的不同取代的环己烷衍生物。特别地,这样的衍生物还可以是可获得的,使用特别地根据现有技术的用于氯胺酮衍生物的已知的制备方法不可获得这样的衍生物。相应地,根据本发明的方法的特征在于高度的灵活性和特别地有助于获得以及提供大量的新颖的环己烷衍生物或,优选地,氯胺酮衍生物。
[0326]
可以在本发明的上下文中进行这样的官能化,例如,通过氢化,特别地在氢气气氛下,优选地在钯催化剂上。
[0327]
进一步地,在本发明的特别优选的实施方式的上下文中,提供的是,将根据本发明的环状2-氨基-1-酮衍生物的6位官能化,特别地羟基化。
[0328]
在此上下文中,非常特别地优选的是,如果氧化性地,优选地使用有机过酸对根据本发明的环状2-氨基-1-酮衍生物的6位进行官能化,特别地羟基化。
[0329]
可以,例如,通过鲁伯特姆(rubottom)氧化实现根据本发明的环状2-氨基-1-酮衍生物的这样的氧化羟基化。在此,特别地使用二异丙基氨基锂,首先将环状2-氨基-1-酮衍生物的羰基基团在α-位去质子化为对应的烯醇化物,其然后与例如三甲基氯硅烷反应以形成硅烯醇醚。替代地,还可能的是,通过催化硅氢加成将环状2-氨基-1-酮衍生物转化为硅烯醇醚。在这种情况下,然后优选提供的是,将铂络合物,特别地pt(0)络合物用作催化剂。特别地,karstedt催化剂作为催化剂,已经证明是很好的。karstedt催化剂是从以下获得的有机铂络合物:使合适的铂化合物,特别地通常六氯铂酸,与含二乙烯基的二硅氧烷反应,以生成铂(0)-1,3-二乙烯基-1,1,3,3-四甲基二硅氧烷络合物。此外,如果使用三烷基硅烷,特别地三乙基硅烷作为硅烷化试剂,对于这种硅烯醇醚形成的替代,已经证明是很好的。
[0330]
随后,特别地使用有机过酸,优选间-氯过苯甲酸,在α-位上进行形式氧化,特别地构成环氧化物,其随后重排以形成α-硅烷氧基酮。最终,硅烷基醚优选裂解成对应的α-羟基酮。
[0331]
在本发明的上下文中,特别有利的是,根据本发明的环状2-氨基-1-酮衍生物的6位的这个优选版本的官能化,特别地羟基化,唯一地产生了顺式构型产物,即,仅仅其中2-氨基基团和引入的6-羟基基团布置在环己烷环的同一侧的那些环状2-氨基-1-酮衍生物。特别地,这也代表了本发明的特别优点,因为不仅可以规避复杂的清洁和分离步骤,而且还特别地以针对性的方式仅合成那些衍生物,其作为氯胺酮和它的代谢物的活性成分或活性类似物是潜在地有效的。特别地,2-氨基基团和6-羟基基团的构型对此是决定性的,其中特别地r,r-构型的衍生物是优选的以及潜在地最有效的。
[0332]
在根据本发明的方法的骨架内,因此特别地提供的是,环状2-氨基-1-酮衍生物,特别地2-氨基环己烷-1-酮衍生物或2-氨基环己烯-1-酮衍生物,优选地2-氨基环己烷-1-酮衍生物,选自通式xvii的衍生物
[0333][0334]
其中
[0335]
r1=芳基、杂芳基;
[0336]
特别地,其中对于r1=芳基,r1选自环状芳基残基,特别地选自萘基、蒽基、菲基残基和通式ii的芳基残基的组
[0337][0338]
其中,各自彼此独立地,
[0339]r11
、r
12
、r
13
=h;
[0340]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、f、cl;
[0341]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷
基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0342]
cooh、-conh2、cosh、cho,特别地cooh、conh2;
[0343]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0344]
苯基、萘基;
[0345]
以及,特别地,其中对于r1=杂芳基,r1选自以下的组:5元环杂芳基,特别地吡咯基、咪唑基、吡唑基、噁唑基、异噁唑基、噻唑基、呋喃基、噻吩基残基和其取代的衍生物,优选吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物,和/或6元杂芳基,特别地吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物,优选吡啶基残基及其取代的衍生物;
[0346]
r2、r3=各自彼此独立地,
[0347]
h;
[0348]c1-c
x-烷基;c
1-c
x-环烷基;
[0349]
或者一起c
1-c
x-环烷基,特别地吡咯烷基、哌啶基、咪唑基、吡啶基残基和它们的取代的衍生物;
[0350]
r4、r5、r6=各自彼此独立地,
[0351]
h;
[0352]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、sh;
[0353]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0354]
cooh、conh2、cosh、cho,特别地cooh、conh2;
[0355]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0356]
其中
[0357]
x=2至20,特别地2至15,优选地2至10;以及
[0358]
y=0、1。
[0359]
在本发明的上下文中,进一步优选的是如果环状2-氨基-1-酮衍生物,特别地2-氨基环己烷-1-酮衍生物或2-氨基环己烯-1-酮衍生物,优选地2-氨基环己烷-1-酮衍生物,选自通式xviii的衍生物
[0360][0361]
其中
[0362]
r1=芳基、杂芳基;
[0363]
其中,对于r1=芳基,r1选自通式iv的芳基残基
[0364][0365]
其中,各自彼此独立地,
[0366]r11
、r
12
、r
13
=h;
[0367]
nh2、oh、f、cl;
[0368]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0369]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0370]
以及其中对于r1=杂芳基,r1选自吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物和/或吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物的组;
[0371]
r2、r3=各自彼此独立地,
[0372]
h;
[0373]c1-c
x-烷基;c
1-c
x-环烷基;
[0374]
r4、r5、r6=各自彼此独立地,
[0375]
h;
[0376]
nh2、oh、sh;
[0377]c1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0378]
其中
[0379]
x=2至10,特别地2至5,优选地2至3;以及
[0380]
y=0、1。
[0381]
此外,在本发明的非常特别优选的实施方式中,可以提供的是,环状2-氨基-1-酮衍生物,特别地2-氨基环己烷-1-酮衍生物或2-氨基环己烯-1-酮衍生物,优选地2-氨基环己烷-1-酮衍生物,选自通式xix的衍生物
[0382][0383]
其中
[0384]
r1=芳基、杂芳基;
[0385]
其中,对于r1=芳基,r1选自通式vi的芳基残基
[0386][0387]
其中,各自彼此独立地,
[0388]r11
、r
12
、r
13
=h;
[0389]
nh2、oh、f、cl;
[0390]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0391]
以及其中对于r1=杂芳基,r1选自噻吩基残基、吡啶基残基以及其取代的衍生物的组;
[0392]
r2、r3=各自彼此独立地,
[0393]
h;
[0394]c1-c
x-烷基;c
1-c
x-环烷基;
[0395]
r4、r5、r6=各自彼此独立地,
[0396]
h;
[0397]
nh2、oh、sh;
[0398]c1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-全氟烷氧基;
[0399]
其中
[0400]
x=2至5,特别地2至3;以及
[0401]
y=0、1。
[0402]
在本发明的上下文中,特别地关于残基r4、r5、r6,因此可以优选地提供的是,环状2-氨基-1-酮衍生物包括环己烯骨架或环己烷骨架。在环状2-氨基-1-酮衍生物包括环己烯骨架的情况下,y=0特别地适用于残基r4、r5。如果环状2-氨基-1-酮衍生物包括环己烷骨架,y=1特别地适用于残基r4、r5。这在上文提及的结构式中由到残基r4、r5的虚线键和虚线双键示出。
[0403]
如开始时已经提及的,使用根据本发明的方法可以实现获得大量的不同的2-氨基-1-酮的衍生物。特别地,在根据本发明的针对性的以及简短的制备方法的范围内,可以以简单的方式和在温和的方法条件下以仅几个步骤从现有的起始化合物生成宽范围的非常不同的环状2-氨基-1-酮衍生物。
[0404]
此外,根据本发明的制备方法的特征在于,特别地进行的反应的高转化率和产率以及高特异性或选择性。因此,特别地从各自反应步骤可靠地产生期望的反应产物,然而几乎没有或根本没有观察到或确定副产物的产生。
[0405]
根据本发明的制备方法的基本部分是初始环化反应,其在环状2-氨基-1-酮衍生物的合成中,首次特别地允许使用空间要求高的取代基,诸如具有任选地进一步空间要求高或富电子的残基的芳基或杂芳基基团。以这种方式,还可以使用根据本发明的方法提供,特别地环状2-氨基-1-酮的完全新颖的衍生物。
[0406]
根据本发明的第二方面的本发明的另外主题,是环状3-烯-2-氧基-1-甲酸衍生物,它的互变异构体、立体异构体、盐或溶液,其中化合物选自通式ix的化合物
[0407][0408]
其中
[0409]
r1=芳基、杂芳基;
[0410]
特别地,其中对于r1=芳基,r1选自环状芳基残基,特别地选自萘基、蒽基、菲基残基和通式ii的芳基残基的组
[0411][0412]
其中,各自彼此独立地,
[0413]r11
、r
12
、r
13
=h;
[0414]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、f、cl;
[0415]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0416]
cooh、-conh2、cosh、cho,特别地cooh、conh2;
[0417]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0418]
苯基、萘基;
[0419]
其中
[0420]
x=2至20,特别地2至15,优选地2至10;
[0421]
以及,特别地,其中对于r1=杂芳基,r1选自以下的组:5元环杂芳基,特别地吡咯基、咪唑基、吡唑基、噁唑基、异噁唑基、噻唑基、呋喃基、噻吩基残基和其取代的衍生物,优选吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物,和/或6元杂芳基,特别地吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物,优选吡啶基残基及其取代的衍生物;
[0422]
pg=ph不稳定的保护基团和/或氧化还原不稳定的保护基团,特别地苄基、对-甲氧基苄基、二甲氧基苄基、烷氧基羰基、三苯基甲基、烷基、烯丙基基团,更优选对-甲氧基苄基、二甲氧基苄基、烷氧基羰基、三苯基甲基基团,更优选对-甲氧基苄基、烷氧基羰基基团;
[0423]r‘
=三烷基硅烷基,特别地三甲基硅烷基、三乙基硅烷基、三异丙基硅烷基、叔丁基二甲基硅烷基、三苯基硅烷基、叔丁基二苯基硅烷基;优选三乙基硅烷基、三异丙基硅烷基、叔丁基二甲基硅烷基。
[0424]
根据本发明的环状3-烯-2-氧基-1-甲酸衍生物的特征特别地在于,它们的高的结构多样性或可变性以及还在于特别地在环己烯主链的双键的区域中进一步进行修饰或官能化的可能性。在这种意义上,根据本发明的环己烯酮衍生物特别地代表在环状2-氨基-1-酮衍生物的合成中的通用中间体,其兼备容易地和廉价地可获得,以及可以在随后反应中以多种方式使用。
[0425]
在此上下文中,根据本发明进一步优选的是如果环状3-烯-2-氧基-1-甲酸衍生物选自通式x的化合物
[0426][0427]
其中
[0428]
r1=芳基、杂芳基
[0429]
其中,对于r1=芳基,r1选自通式iv的芳基残基
[0430][0431]
其中,各自彼此独立地,
[0432]r11
、r
12
、r
13
=h;
[0433]
nh2、oh、f、cl;
[0434]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0435]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0436]
以及
[0437]
x=2至10,特别地2至5,优选地2至3;
[0438]
以及其中对于r1=杂芳基,r1选自吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物和/或吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物的组;
[0439]
pg=ph不稳定的保护基团和/或氧化还原不稳定的保护基团,特别地对-甲氧基苄基、二甲氧基苄基、烷氧基羰基、三苯基甲基基团;
[0440]r‘
=三乙基硅烷基、三异丙基硅烷基、叔丁基二甲基硅烷基。
[0441]
在根据本发明的方法的更优选的实施方式中,还可以提供的是,环状3-烯-2-氧基-1-甲酸衍生物选自通式xi的化合物
[0442][0443]
其中
[0444]
r1=芳基、杂芳基
[0445]
其中,对于r1=芳基,r1选自通式vi的芳基残基
[0446][0447]
其中,各自彼此独立地,
[0448]r11
、r
12
、r
13
=h;
[0449]
nh2、oh、f、cl;
[0450]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0451]
以及
[0452]
x=2至5,特别地2至3;
[0453]
以及其中对于r1=杂芳基,r1选自噻吩基残基、吡啶基残基以及其取代的衍生物的组;
[0454]
pg=ph不稳定的保护基团和/或氧化还原不稳定的保护基团,特别地
[0455]
对-甲氧基苄基、二甲氧基苄基、烷氧基羰基;
[0456]r‘
=三乙基硅烷基、三异丙基硅烷基、叔丁基二甲基硅烷基。
[0457]
对于根据本发明的环状3-烯-2-氧基-1-甲酸衍生物的另外细节,可以参考上文对
根据本发明的方法的解释,其类似地适用于环状3-烯-2-氧基-1-甲酸衍生物。
[0458]
再次,根据本发明的第三方面的本发明的另外主题是,一种用于生产环状3-烯-2-氧基-1-甲酸衍生物,特别地根据本发明的环状3-烯-2-氧基-1-甲酸衍生物的方法,其中使芳族丙烯酸衍生物,特别地α-碳芳族取代的丙烯酸衍生物,在环化反应中与1,3-丁二烯醇衍生物反应。
[0459]
根据本发明,在此优选提供的是,环化反应是环加成,特别地[4 2]环加成,优选地第尔斯-阿尔德反应。
[0460]
对于进行根据本发明的方法,如果芳族丙烯酸衍生物选自通式i的化合物,还已经证明是很好的
[0461][0462]
其中
[0463]
r1=芳基、杂芳基
[0464]
特别地,其中对于r1=芳基,r1选自环状芳基残基,特别地选自萘基、蒽基、菲基残基和通式ii的芳基残基的组
[0465][0466]
其中,各自彼此独立地,
[0467]r11
、r
12
、r
13
=h;
[0468]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、f、cl;
[0469]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0470]
cooh、-conh2、cosh、cho,特别地cooh、conh2;
[0471]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0472]
苯基、萘基;
[0473]
其中
[0474]
x=2至20,特别地2至15,优选地2至10;
[0475]
以及,特别地,其中对于r1=杂芳基,r1选自以下的组:5元环杂芳基,特别地吡咯基、咪唑基、吡唑基、噁唑基、异噁唑基、噻唑基、呋喃基、噻吩基残基和其取代的衍生物,优选吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物,和/或6元杂芳基,特别地吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物,优选吡啶基残基及其取代的衍生物;
[0476]
pg=ph不稳定的保护基团和/或氧化还原不稳定的保护基团,
[0477]
特别地苄基、对-甲氧基苄基、二甲氧基苄基、烷氧基羰基、三苯基甲基、烷基、烯丙基基团,更优选对-甲氧基苄基、二甲氧基苄基、烷氧基羰基、三苯基甲基基团,更优选对-甲
氧基苄基、烷氧基羰基基团。
[0478]
在本发明的上下文中,如果芳族丙烯酸衍生物选自通式iii的化合物,获得特别良好的结果
[0479][0480]
其中
[0481]
r1=芳基、杂芳基;
[0482]
其中,对于r1=芳基,r1选自通式iv的芳基残基
[0483][0484]
其中,各自彼此独立地,
[0485]r11
、r
12
、r
13
=h;
[0486]
nh2、oh、f、cl;
[0487]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0488]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0489]
以及
[0490]
x=2至10,特别地2至5,优选地2至3;
[0491]
以及其中对于r1=杂芳基,r1选自吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物和/或吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物的组;
[0492]
pg=ph不稳定的保护基团和/或氧化还原不稳定的保护基团,特别地对-甲氧基苄基、二甲氧基苄基、烷氧基羰基、三苯基甲基基团。
[0493]
根据本发明的非常特别优选的实施方式,优选的是如果芳族丙烯酸衍生物选自通式v的化合物
[0494][0495]
其中
[0496]
r1=芳基、杂芳基;
[0497]
其中,对于r1=芳基,r1选自通式vi的芳基残基
[0498][0499]
其中,各自彼此独立地,
[0500]r11
、r
12
、r
13
=h;
[0501]
nh2、oh、f、cl;
[0502]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0503]
以及
[0504]
x=2至5,特别地2至3;
[0505]
以及其中对于r1=杂芳基,r1选自噻吩基残基、吡啶基残基以及其取代的衍生物的组;
[0506]
pg=ph不稳定的保护基团和/或氧化还原不稳定的保护基团,特别地对-甲氧基苄基、二甲氧基苄基、烷氧基羰基。
[0507]
关于1,3-丁二烯醇衍生物,在本发明的上下文中,如果1,3-丁二烯醇衍生物选自富电子1,3-丁二烯醇衍生物,特别地选自通式vii的1,3-丁二烯醇衍生物,已经证明是有利的
[0508][0509]
其中
[0510]r‘
=c
1-c
x-烷基、c
1-c
x-乙烯基、c
1-c
x-烯丙基;
[0511]
苯基、苄基;
[0512]
三烷基硅烷基;
[0513]
以及
[0514]
x=2至10。
[0515]
特别优选地,在本发明的上下文中,1,3-丁二烯醇衍生物选自通式viii的丁二烯氧基硅烷
[0516][0517]
其中
[0518]r‘
=三烷基硅烷基;
[0519]
三烷基硅烷基,特别地三甲基硅烷基、三乙基硅烷基、三异丙基硅烷基、叔丁基二甲基硅烷基、三苯基硅烷基、叔丁基二苯基硅烷基;优选三乙基硅烷基、三异丙基硅烷基、叔丁基二甲基硅烷基。
[0520]
关于反应条件或在环化反应中的两种上文提及的起始化合物的反应期间的条件,这些对于本领域技术人员通常是已知的。
[0521]
然而,对于根据本发明的方法,如果芳族丙烯酸衍生物与1,3-丁二烯醇衍生物的反应是以在0.5:1至1:20、特别地1:1至1:15、优选地1:2至1:10的范围内的芳族丙烯酸衍生物与1,3-丁二烯醇衍生物的比率进行,获得特别良好的结果。
[0522]
此外,如果反应是在惰性溶剂中进行的,特别地环状的,特别地芳族的、烃,优选四氢呋喃、二甲苯或其混合物的溶剂中进行的,已经证明是有利的。
[0523]
同样地,在根据本发明的方法中如果反应在供热下进行,特别地在50至250℃、优选75至200℃、优选100至175℃的范围内的温度下进行,已经证明是很好的。
[0524]
此外,优选地提供的是,反应在5至300min,特别地10至250min,优选地15至200min
的时间段内进行。
[0525]
最后但同样重要的是,在本发明的上下文中,如果反应是在自由基清除剂的存在下,特别地在二羟基苯的存在下,优选地在氢醌的存在下进行,获得特别良好的结果。
[0526]
根据本发明的用于制备环状3-烯-2-氧基-1-甲酸衍生物的方法的特征特别地在于,可靠地实现高转化率,其中同时,可以观察到高度选择性的产生期望的反应产物。这是特别地令人惊讶的,因为根据本发明的方法还优选涉及使用富电子或空间倾向受阻的二烯,其反应性被相应地降低。然而实现高转化率和良好的产率的事实特别地是因为根据本发明的起始化合物的有利的组合,其在环状3-烯-2-氧基-1-甲酸衍生物和基于此的环状2-氨基-1-酮衍生物的合成的上下文中首先进行描述。
[0527]
对于根据本发明的方法的另外细节,可以参考上文对本发明的其他方面的解释,其相应地适用于根据本发明的方法。
[0528]
根据本发明的第四方面的本发明的另外主题,是二环氨基甲酸酯衍生物,它的互变异构体、立体异构体、盐或溶液,其特征在于,化合物选自通式xiii的化合物,
[0529][0530]
其中
[0531]
r1=芳基、杂芳基;
[0532]
特别地,其中对于r1=芳基,r1选自环状芳基残基,特别地选自萘基、蒽基、菲基残基和通式ii的芳基残基的组
[0533][0534]
其中,各自彼此独立地,
[0535]r11
、r
12
、r
13
=h;
[0536]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、f、cl;
[0537]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0538]
cooh、-conh2、cosh、cho,特别地cooh、conh2;
[0539]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0540]
苯基、萘基;
[0541]
或者,其中对于r1=杂芳基,r1选自以下的组:5元环杂芳基,特别地吡咯基、咪唑基、吡唑基、噁唑基、异噁唑基、噻唑基、呋喃基、噻吩基残基和其取代的衍生物,优选吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物,和/或6元杂芳基,特别地吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物,优选吡啶基残基及其取代的衍生物;
[0542]
r2、r3=各自彼此独立地,
[0543]
h;
[0544]c1-c
x-烷基;c
1-c
x-环烷基;
[0545]
r4、r5、r6=各自彼此独立地,
[0546]
h;
[0547]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、sh;
[0548]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0549]
cooh、conh2、cosh、cho,特别地cooh、conh2;
[0550]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0551]
其中
[0552]
x=2至20,特别地2至15,优选地2至10;以及
[0553]
y=0、1。
[0554]
与环状3-烯-2-氧基-1-甲酸衍生物一样,根据本发明的二环氨基甲酸酯衍生物的特征特别地在于以下事实,它包括用于进一步对分子结构进行多样化地修饰或官能化的特定潜能。
[0555]
优选地,从二环氨基甲酸酯衍生物开始,可以以几个并且不复杂的步骤提供宽范围的目标化合物。特别地,这些目标化合物可以选自氯胺酮或它的代谢物去甲氯胺酮和6-羟基去甲氯胺酮的衍生物。
[0556]
因此,根据本发明的二环氨基甲酸酯衍生物,以及根据本发明的环状3-烯-2-氧基-1-甲酸衍生物,代表了在环状2-氨基-1-酮衍生物的轻易可得的合成中的核心中间步骤。例如,从本发明的二环氨基甲酸酯衍生物开始,可以制备基于环己烯和基于环己烷的目标化合物两者,其可以进一步包括在环己烷环上不同的取代模式。
[0557]
在本发明的上下文中,特别优选的是如果化合物,即,二环氨基甲酸酯衍生物选自通式xiv的化合物
[0558][0559]
其中
[0560]
r1=芳基、杂芳基;
[0561]
其中,对于r1=芳基,r1选自通式iv的芳基残基
[0562][0563]
其中,各自彼此独立地,
[0564]r11
、r
12
、r
13
=h;
[0565]
nh2、oh、f、cl;
[0566]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0567]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0568]
以及其中对于r1=杂芳基,r1选自吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物和/或吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物的组;
[0569]
r4、r5、r6=各自彼此独立地,
[0570]
h;
[0571]
nh2、oh、sh;
[0572]c1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0573]
其中
[0574]
x=2至10,特别地2至5,优选地2至3;以及
[0575]
y=0、1。
[0576]
此外,根据本发明的更优选的实施方式,可以提供的是,二环氨基甲酸酯衍生物选自通式xv的衍生物
[0577][0578]
其中
[0579]
r1=芳基、杂芳基;
[0580]
其中,对于r1=芳基,r1选自通式vi的芳基残基
[0581][0582]
其中,各自彼此独立地,
[0583]r11
、r
12
、r
13
=h;
[0584]
nh2、oh、f、cl;
[0585]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0586]
以及其中对于r1=杂芳基,r1选自噻吩基残基、吡啶基残基以及其取代的衍生物的组;
[0587]
r4、r5、r6=各自彼此独立地,
[0588]
h;
[0589]
nh2、oh、sh;
[0590]c1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-全氟烷氧基;
[0591]
其中
[0592]
x=2至5,特别地2至3;以及
[0593]
y=0、1。
[0594]
对于根据本发明的二环氨基甲酸酯衍生物的另外细节,可以参考上文对本发明的其他方面的解释,其类似地适用于二环氨基甲酸酯衍生物。
[0595]
此外,根据本发明的第五方面的本发明的另外主题,是用于制备二环氨基甲酸酯衍生物,特别地根据本发明的二环氨基甲酸酯衍生物的方法,其中环状3-烯-2-氧基-1-甲酸衍生物,特别地根据本发明的环状3-烯-2-氧基-1-甲酸衍生物,与叠氮化物试剂进行叠氮化。
[0596]
在根据本发明的方法中,在此上下文中如果优选在还原和/或碱性或酸性条件下去除保护基团(pg)和/或残基r’,特别地保护基团(pg)和残基r’,已经证明是很好的。
[0597]
在这种意义上,根据本发明优选的是,如果保护基团(pg)和/或残基r’,特别地保护基团(pg)和残基r’的去除,氢解地,即通过在金属催化剂上在氢气的存在下还原,和/或在碱性或酸性条件下,优选地在氟离子的存在下,例如使用四丁基氟化铵(tbaf)进行。同样地,然而,本发明还可以使用本领域技术人员通常使用的用于去除保护基团的方案。
[0598]
在本发明的更优选实施方式的上下文中,优选的是如果在随后将残基r'在碱性或酸性条件下特别地在氟离子的存在下去除之前,氢解地,即通过在金属催化剂上在氢气的存在下还原,和/或在碱性或酸性条件下首先去除保护基团(pg)。在此上下文中,还可以优选的是,在保护基团(pg)的去除过程中或在残基r’的去除之前,使环己烯骨架,特别地环己烯骨架的双键官能化。
[0599]
关于叠氮化条件,此外已经证明有利的是,在本发明的上下文中,如果使用选自以下的组的试剂进行叠氮化:无机叠氮化物,特别地叠氮化钠,和/或有机叠氮化物,特别地磺酰叠氮、磷酰叠氮,优选地对甲苯磺酰叠氮、二苯基磷酰叠氮,或其混合物。
[0600]
在此,特别地,在根据本发明的方法中,如果用于叠氮化的试剂是以过量1至20倍,特别地过量1至10倍,优选地过量1.5至7倍使用,在每种情况下均基于所使用的环状3-烯-2-氧基-1-甲酸衍生物的量,获得良好的结果。
[0601]
此外,对于根据本发明的方法,如果在碱,特别地有机碱,优选地氮碱,更优选地叔胺,更优选地三乙胺的存在下进行叠氮化,获得特别良好的结果。
[0602]
替代地,在本发明的范围内可以使用其他碱,特别地氮碱,其中合适的碱,特别地氮碱本身为本领域技术人员熟悉。在本发明的上下文中特别地优选的通常是,那些仅包括低亲核特性的碱,如例如对于更高取代的胺的情况。
[0603]
同样地,如果叠氮化是在惰性溶剂中进行的,特别地环状的,特别地芳族的烃,优选甲苯的溶剂中进行的,已经证明是有利的。
[0604]
此外,根据本发明,优选的是如果叠氮化在供热下进行,特别地在40至200℃、优选50至150℃、优选60至125℃的范围内的温度下进行。
[0605]
最后,在本发明的上下文中,优选提供的是,在1小时至48小时、特别地1.5至36小时、优选地1.5至24小时的时间段内进行酸化。
[0606]
特别地,在本发明的上下文中还提供的是,将羧酸叠氮化物环化为氨基甲酸酯,特别地通过重排为相应地异氰酸酯。
[0607]
如果,另外,期望进一步修饰或官能化,在本发明的上下文中如果在叠氮化之前的反应步骤中,将环己烯骨架,特别地环己烯骨架的双键官能化,优选地产生化合物xii,已经
证明是特别有用的
[0608][0609]
其中残基r4、r5、r6,特别地其中所述残基r4、r5、r6各自彼此独立地,选自以下的组:
[0610]
r4、r5、r6=h;
[0611]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、sh;
[0612]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0613]
cooh、conh2、cosh、cho,特别地cooh、conh2;
[0614]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0615]
其中
[0616]
x=2至20,特别地2至15,优选地2至10,更优选地2至5,更优选地2至3;以及
[0617]
y=0、1,其中对于残基r4、r5、r6中的至少一个,y=1。
[0618]
对于根据本发明的方法的另外细节,可以参考上文对本发明的其他方面的解释,其类似地适用于根据本发明的方法。
[0619]
根据本发明的第六方面的本发明的另外主题,是环状2-氨基-1-酮衍生物,特别地2-氨基环己烷-1-酮衍生物或2-氨基环己烯-1-酮衍生物,优选地2-氨基环己烷-1-酮衍生物,它的互变异构体、立体异构体、盐或溶液,其中化合物选自通式xvii的化合物,
[0620][0621]
其中
[0622]
r1=芳基、杂芳基;
[0623]
特别地,其中对于r1=芳基,r1选自环状芳基残基,特别地选自萘基、蒽基、菲基残基和通式ii的芳基残基的组
[0624][0625]
其中,各自彼此独立地,
[0626]r11
、r
12
、r
13
=h;
[0627]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、f、cl;
[0628]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷
基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0629]
cooh、-conh2、cosh、cho,特别地cooh、conh2;
[0630]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0631]
苯基、萘基;
[0632]
以及,特别地,其中对于r1=杂芳基,r1选自以下的组:5元环杂芳基,特别地吡咯基、咪唑基、吡唑基、噁唑基、异噁唑基、噻唑基、呋喃基、噻吩基残基和其取代的衍生物,优选吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物,和/或6元杂芳基,特别地吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物,优选吡啶基残基及其取代的衍生物;
[0633]
r2、r3=各自彼此独立地,
[0634]
h;
[0635]c1-c
x-烷基;c
1-c
x-环烷基;
[0636]
或者一起c
1-c
x-环烷基,特别地吡咯烷基、哌啶基、咪唑基、吡啶基残基和它们的取代的衍生物;
[0637]
r4、r5、r6=各自彼此独立地,
[0638]
h;
[0639]
nh2、oh、sh、f、cl、br、no2,特别地nh2、oh、sh;
[0640]c1-c
x-单烷基氨基、c
1-c
x-二烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基硫基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基,特别地c
1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0641]
cooh、conh2、cosh、cho,特别地cooh、conh2;
[0642]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基)、cos(c
1-c
x-烷基)、c(o)(c
1-c
x-烷基),特别地coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0643]
其中
[0644]
x=2至20,特别地2至15,优选地2至10;以及
[0645]
y=0、1。
[0646]
特别地,在本发明的上下文中,优选的是如果化合物,即环状2-氨基-1-酮衍生物选自通式xviii的化合物,
[0647][0648]
其中
[0649]
r1=芳基、杂芳基;
[0650]
其中,对于r1=芳基,r1选自通式iv的芳基残基
[0651]
[0652]
其中,各自彼此独立地,
[0653]r11
、r
12
、r
13
=h;
[0654]
nh2、oh、f、cl;
[0655]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0656]
coo(c
1-c
x-烷基)、conh(c
1-c
x-烷基);
[0657]
以及其中对于r1=杂芳基,r1选自吡啶基、吡嗪基、哒嗪基、嘧啶基残基及其取代的衍生物和/或吡咯基、咪唑基、吡唑基、噻吩基残基及其取代的衍生物的组;
[0658]
r2、r3=各自彼此独立地,
[0659]
h;
[0660]c1-c
x-烷基;c
1-c
x-环烷基;
[0661]
r4、r5、r6=各自彼此独立地,
[0662]
h;
[0663]
nh2、oh、sh;
[0664]c1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0665]
其中
[0666]
x=2至10,特别地2至5,优选地2至3;以及
[0667]
y=0、1。
[0668]
此外,在本发明的非常特别优选的实施方式中,可以提供的是,环状2-氨基-1-酮衍生物,特别地2-氨基环己烷-1-酮衍生物或2-氨基环己烯-1-酮衍生物,优选地2-氨基环己烷-1-酮衍生物,选自通式xix的衍生物
[0669][0670]
其中
[0671]
r1=芳基、杂芳基;
[0672]
其中,对于r1=芳基,r1选自通式vi的芳基残基
[0673][0674]
其中,各自彼此独立地,
[0675]r11
、r
12
、r
13
=h;
[0676]
nh2、oh、f、cl;
[0677]c1-c
x-烷氧基、c
1-c
x-烷基、c
1-c
x-全氟烷氧基、c
1-c
x-全氟烷基;
[0678]
以及其中对于r1=杂芳基,r1选自噻吩基残基、吡啶基残基以及其取代的衍生物的组;
[0679]
r2、r3=各自彼此独立地,
[0680]
h;
[0681]c1-c
x-烷基;c
1-c
x-环烷基;
[0682]
r4、r5、r6=各自彼此独立地,
[0683]
h;
[0684]
nh2、oh、sh;
[0685]c1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-全氟烷氧基;
[0686]
其中
[0687]
x=2至5,特别地2至3;以及
[0688]
y=0、1。
[0689]
在本发明的上下文中,现在优选的是如果化合物,即环状2-氨基-1-酮衍生物选自以下的组
[0690]
[0691]
[0692][0693]
其中
[0694]
r2、r3=各自彼此独立地,
[0695]
h;
[0696]c1-c
x-烷基;c
1-c
x-环烷基;
[0697]
r4=h;
[0698]
nh2、oh、sh;
[0699]c1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-全氟烷氧基;
[0700]
其中
[0701]
x=2至10,特别地2至5,优选地2至3。
[0702]
甚至更优选地,化合物,即环状2-氨基-1-酮衍生物选自以下的组
[0703]
[0704]
[0705][0706]
其中
[0707]
r2、r3=各自彼此独立地,
[0708]
h;
[0709]c1-c
x-烷基;c
1-c
x-环烷基;
[0710]
r4=h;
[0711]
nh2、oh、sh;
[0712]c1-c
x-单烷基氨基、c
1-c
x-烷氧基、c
1-c
x-全氟烷氧基;
[0713]
其中
[0714]
x=2至10,特别地2至5,优选地2至3。
[0715]
对于根据本发明的环状2-氨基-1-酮衍生物的另外细节,可以参考上文对本发明的其他方面的解释,其类似地适用于环状2-氨基-1-酮衍生物。
[0716]
根据本发明的第七方面的本发明的另外主题,是根据本发明的环状2-氨基-1-酮衍生物,特别地2-氨基环己烷-1-酮衍生物或2-氨基环己烯-1-酮衍生物,优选地2-氨基环己烷-1-酮衍生物,它们的互变异构体、立体异构体、盐或溶液用于在刺激和/或恢复神经元的神经元可塑性中的用途。
[0717]
在这种意义上,本发明的主题还是根据本发明的或通过根据本发明的方法可获得的环状2-氨基-1-酮衍生物,特别地2-氨基环己烷-1-酮衍生物或2-氨基环己烯-1-酮衍生物,优选地2-氨基环己烷-1-酮衍生物,它们的互变异构体、立体异构体、盐或溶液,用于刺激和/或恢复神经元的神经元可塑性的用途。
[0718]
对于根据本发明的环状2-氨基-1-酮衍生物的用途的另外细节,可以参考上文对本发明的其他方面的解释,其类似地适用于根据本发明的用途。
[0719]
此外,根据本发明的第八方面的本发明的另外主题,是根据本发明的环状2-氨基-1-酮衍生物,特别地2-氨基环己烷-1-酮衍生物或2-氨基环己烯-1-酮衍生物,优选地2-氨基环己烷-1-酮衍生物,它们的互变异构体、立体异构体、盐或溶液,用于在刺激和/或恢复神经元的突触可塑性中的用途。
[0720]
在这种意义上,本发明的主题还是根据本发明的或通过根据本发明的方法可获得的环状2-氨基-1-酮衍生物,特别地2-氨基环己烷-1-酮衍生物或2-氨基环己烯-1-酮衍生物,优选地2-氨基环己烷-1-酮衍生物,它们的互变异构体、立体异构体、盐或溶液,用于刺激和/或恢复神经元的突触可塑性的用途。
[0721]
对于根据本发明的环状2-氨基-1-酮衍生物的用途的另外细节,可以参考上文对本发明的其他方面的解释,其类似地适用于根据本发明的用途。
[0722]
根据本发明的第九方面的本发明的另外主题,是根据本发明的环状2-氨基-1-酮衍生物,特别地2-氨基环己烷-1-酮衍生物或2-氨基环己烯-1-酮衍生物,优选地2-氨基环己烷-1-酮衍生物,它们的互变异构体、立体异构体、盐或溶液,用于作为药物,特别地在预防性或治疗性治疗人或动物身体的疾病,优选地神经退行性疾病,特别地痴呆、阿尔茨海默病、帕金森病,和/或精神疾病,特别地抑郁中的用途。
[0723]
特别地,对于根据本发明的环状2-氨基-1-酮衍生物已经观察到,它们能够引发可以有助于神经细胞的突触可塑性的恢复的过程。本发明的化合物以有利的方式有助于此,特别地通过支持神经细胞的树突的形成和进一步的分支。
[0724]
以这种方式,如果不逆转,可以最终使神经系统变性过程停止,使得特别地基于根据本发明的化合物可能够有效地治疗基础性神经退行性疾病。
[0725]
此外,根据本发明的环状2-氨基-1-酮衍生物具有在卒中、创伤或缺氧脑损伤之后的康复期间有效支持神经细胞再生的潜能。
[0726]
对于根据本发明的环状2-氨基-1-酮衍生物的用途的另外细节,可以参考上文对本发明的其他方面的解释,其类似地适用于用途。
[0727]
最终,根据本发明的第十方面的本发明的另外主题,是药物组合物,特别地药品或药物,包括根据本发明的环状2-氨基-1-酮衍生物,特别地2-氨基环己烷-1-酮衍生物或2-氨基环己烯-1-酮衍生物,优选地2-氨基环己烷-1-酮衍生物,优选地用于在预防性或治疗性治疗人或动物身体的疾病,特别地神经退行性疾病,特别地痴呆和/或神经认知疾患,其由以下导致的:阿尔茨海默病、帕金森病、皮克病、颅脑创伤、亨廷顿病、路易体,和/或精神疾病,特别地抑郁中的用途。
[0728]
对于药物组合物的另外细节,可以参考上文对本发明的其他方面的解释,其类似地适用于组合物。
[0729]
本发明的主题将参照实施方式的实施例以非限制的方式进行说明。
[0730]
示例性实施方式:
[0731]
根据本发明的制备方法以及特别地根据本发明的制备方法的单独的反应步骤,通过以下实施方式进一步说明。
[0732]
总体上,因此根据本发明优选地提供的是,用于制备环状2-氨基-1-酮衍生物的方
法根据以下通用反应方案进行,其中残基r'、pg、r1、r2、r3、r
4y
、r
5y
和r
6y
的定义参照上文对本发明的单独方面的解释:
[0733][0734]
1.环状3-烯-2-氧基-1-甲酸衍生物:
[0735]
1.1.通用程序
[0736][0737]
向芳族丙烯酸衍生物在二甲苯和/或thf中的溶液中,在室温下加入过量2至8倍的1,3-丁二烯醇衍生物。然后在75℃至200℃下将产生的混合物搅拌5至250分钟。
[0738]
这个时间后,通过在真空中去除溶剂终止反应。通过柱色谱法在硅胶上使用环己烷和乙酸乙酯的溶剂混合物对获得的残余物进行纯化。
[0739]
1.2.特定实施例:
[0740]
(a)合成rac-4-甲氧基苄基-6-((三乙基硅烷基)氧基)-1,2,3,6-四氢-[1,1'-联苯基]-1-甲酸酯
[0741]
向134mg的4-甲氧基苄基-2-苯基丙烯酸酯(0.50mmol,1.00eq.)在0.5ml的邻-二甲苯中的溶液中,加入553mg的(e)-(丁-1,3-二烯-1-基氧基)三乙基硅烷(3.00mmol,6.00eq.)和一小称量勺尖(spatula tip)的氢醌。在微波中在140℃下加热反应混合物20分钟。
[0742]
减压去除溶剂之后,通过柱色谱法在硅胶上(0
→
10%乙酸乙酯与环己烷)纯化残余物。
[0743]
获得180mg(0.40mmol,80%)的rac-4-甲氧基苄基-6-((三乙基硅烷基)氧基)-1,2,3,6-四氢-[1,1'-联苯基]-1-甲酸酯的内型/外型混合物(1:1),呈无色油。
[0744]
(b)合成2'-甲氧基-6-((三乙基硅烷基)氧基)-3,6-二氢-[1,1'-联苯基]-1(2h)-甲酸rac-甲酯
[0745]
向96mg的2-(2'-甲氧基)苯基丙烯酸甲酯(0.50mmol,1.00eq.)在0.5ml的邻-二甲苯中的溶液中,加入368mg的(e)-(丁-1,3-二烯-1-基氧基)三乙基硅烷(2.00mmol,4.00eq.)和一小称量勺尖的氢醌。在微波烘箱中在140℃下将反应混合物加热6h。
[0746]
减压去除溶剂之后,通过柱色谱法在硅胶上(0
→
10%乙酸乙酯与环己烷)纯化残余物。
[0747]
获得117mg(0.31mmol,62%)的2
’‑
甲氧基-6-((三乙基硅烷基)氧基)-3,6-二氢-[1,1'-联苯基]-1(2h)-甲酸rac-甲酯的内型/外型混合物(9:1),呈无色油。
[0748]
(c)合成rac-4-甲氧基苄基-6-((三乙基硅烷基)氧基)-2'-(三氟甲氧基)-3,6-二氢-[1,1'-联苯基]-1(2h)-甲酸酯
[0749]
向96mg的4-甲氧基苄基-2-(2'-三氟甲氧基)苯基丙烯酸酯(0.26mmol,1.00eq.)在0.3ml的邻-二甲苯中的溶液中,加入167mg的(e)-(丁-1,3-二烯-1-基氧基)三乙基硅烷(2.00mmol,4.00eq.)和一小称量勺尖的氢醌。在微波烘箱中在140℃下将反应混合物加热7h。
[0750]
减压去除溶剂之后,通过柱色谱法在硅胶上(0
→
10%乙酸乙酯与环己烷)纯化残余物。
[0751]
获得60mg(0.11mmol,42%)的rac-4-甲氧基苄基-6-((三乙基硅烷基)氧基)-2'-(三氟甲氧基)-3,6-二氢-[1,1'-联苯基]-1(2h)-甲酸酯的内型-/外型混合物(3:1),呈无色油。
[0752]
1.3.制备的环状3-烯-2-氧基-1-甲酸衍生物的概述和表征
[0753]
下表给出了根据本发明制备的3-烯-2-氧基-1-甲酸衍生物以及在每种情况下获得的产率的概述。
[0754]
[0755]
[0756]
[0757][0758]
2.二环氨基甲酸酯衍生物的合成:
[0759]
2.1.通用程序
[0760][0761]
首先在标准条件下,将从第一反应步骤中获得的环状3-烯-2-氧基-1-甲酸衍生物去保护或去封端,即根据通用标准方案,例如在还原或酸性条件下去除pg和r'残基。然后,通过柱色谱法在硅胶上使用环己烷和乙酸乙酯的溶剂混合物对获得的粗混合物进行纯化。
[0762]
在第二反应步骤的下一个子步骤中,将去保护或去封端的环状3-烯-2-氧基-1-甲酸衍生物溶解在thf或甲苯中,以及加入过量1至5倍的三乙胺和过量1至5倍的二苯基磷酰基叠氮化物。在40至125℃下将产生的反应混合物搅拌1至36小时。
[0763]
通过加入水终止反应,以及通过柱色谱法在硅胶上使用环己烷和乙酸乙酯的溶剂混合物将在水性后处理和萃取之后获得的残余物进行纯化。
[0764]
另外,在制备二环氨基甲酸酯的过程中,还可以对环己烯骨架的双键进行修饰。例如,在保护基团pg的还原去除过程中还可以实现对双键的氢化。为了这个目的,特别地在钯催化剂的存在下在氢气气氛下进行环状3-烯-2-氧基-1-甲酸衍生物的反应是合适的。然而,在这方面的反应程序通常还是已知的。
[0765]
其他修饰变体,诸如在例如用杂原子(基团)诸如特别地卤素或羟基基团的取代反应的过程中双键的打开,也可以优选在制备二环氨基甲酸酯的过程中进行。
[0766]
2.2.特定实施例:3a-苯基六氢苯并[d]噁唑-2(3h)-酮的合成
[0767]
将180mg(0.40mmol,1.00eq.)的6-((三乙基硅烷基)氧基)-1,2,3,6-四氢-[1,1'-联苯基]-1-甲酸rac-4-甲氧基苄酯溶解在4ml的甲醇中,将活性炭上的一称量勺尖的钯(10%)加入以及在室温下在氢气气氛下搅拌反应混合物17小时。
[0768]
然后过滤出催化剂以及在真空中从溶剂中释放出滤液。将残余物溶解在4ml的二氯甲烷中,使溶液冷却到0℃以及将308μl三氟乙酸(4.00mmol,10.00eq.)加入。在室温下搅拌4h后,使用旋转蒸发器去除溶剂以及通过柱色谱法在硅胶上(0
→
40%乙酸乙酯与环己烷)纯化残余物,产生81mg(0.37mmol,92%)的2-羟基-1-苯基环己烷甲酸的内型/外型混合物(3:2),呈无色油。
[0769]
在其他方法中,向溶解在6.5ml的甲苯中的163mg的2-羟基-1-苯基环己烷甲酸(0.74mmol,1.00eq.)中,加入308μl的三乙胺(2.22mmol,3.00eq.)和479μl的二苯基磷酰基叠氮化物(2.22mmol,3.00eq.)。首先将溶液加热到80℃持续2小时,其中观察到气体释放,以及然后在室温下搅拌过夜。
[0770]
向反应混合物中,加入10ml乙酸乙酯和10ml水,以及用乙酸乙酯(3x10 ml)萃取。将合并的有机相用饱和nahco3溶液和饱和nacl溶液洗涤,经na2so4干燥,过滤并且在真空中浓缩。通过柱色谱法在硅胶上(0
→
60%乙酸乙酯与环己烷)纯化残余物,产生115mg的内型/外型混合物(1:1)的3a-苯基六氢苯并[d]噁唑-2(3h)-酮(0.53mmol,72%)的,呈淡黄油。
[0771]
2.3.制备的二环氨基甲酸酯衍生物的概述和表征
[0772]
下表提供了根据本发明制备的二环氨基甲酸酯衍生物以及在每种情况下获得的产率的概述。
[0773][0774]
3.环状2-氨基-1-酮衍生物的合成:
[0775]
3.1.通用程序
[0776][0777]
将来自之前的反应步骤的二环氨基甲酸酯衍生物溶解在thf或二噁烷中,以及加入过量3至20倍的锂盐,例如氢化铝锂或氢氧化锂。然后在25至180℃下将获得的反应混合物搅拌15至24小时。
[0778]
这个时间后,通过加入水或稀释的盐酸终止反应,因此将反应混合物水性地后处理以及在水相分离之后将粗产物从剩余的有机溶剂中释放出。
[0779]
随后,将获得的残余物溶解在丙酮中并且在冷却下将过量3至8倍的在浓硫酸中的
氧化铬(vi)加入。将获得的反应混合物在-10至40℃下搅拌1至5小时。最后通过加入异丙醇终止反应,因此进行水性后处理并且最终通过萃取分离产物。
[0780]
3.2.特定实施例:2-(甲基氨基)-2-苯基环己酮的合成
[0781]
向115mg的3a-苯基六氢苯并[d]噁唑-2(3h)-酮(0.53mmol,1.00eq.)在2.5ml的thf中的溶液中,在0℃下分批加入100mg氢化铝锂(2.60mmol,5.00eq.)。在室温下将浅灰色悬浮液搅拌一小时,将混合物回流17小时。
[0782]
冷却至室温后,将悬浮液用3ml的醚稀释以及冷却至0℃。然后逐滴加入100μl的水、100μl的15%naoh水性溶液和另外300μl的水。在室温下搅拌15分钟后,加入无水mgso4以及将混合物再搅拌15分钟。然后过滤出固体以及减压下从溶剂中释放出滤液。
[0783]
将获得的残余物溶解在10.6ml丙酮中以及在0℃下与cro3在h2so4中的1.1ml的2m溶液(2.20mmol,4.15eq.)逐滴混合。然后将反应在室温下搅拌2小时以及然后通过加入20ml的异丙醇进行终止。将溶液用2m naoh调整到ph 12,用醚(3x10 ml)萃取水相,将合并的有机相经na2so4干燥,过滤以及使用旋转蒸发器从溶剂中释放,产生106mg(0.52mmol,99%)的2-(甲基氨基)-2-苯基环己酮,呈黄色油。
[0784]
3.3.制备的环状2-氨基-1-酮衍生物的概述和表征
[0785]
在下表中,进而给出了根据本发明制备的环状2-氨基-1-酮衍生物以及在每种情况下获得的产率的概述。
[0786][0787]
[0788]
4.环状2-氨基-1-酮衍生物的环己烷主链的修饰
[0789]
可以以多种方式对根据本发明制备的环状2-氨基-1-酮衍生物进行进一步的修饰。特别地,在此可以提供具有另外的官能团或另外残基,诸如烷基残基的环己烷骨架。
[0790]
在此上下文中,特别地优选的是环己烷骨架的6位的羟基化,其可以例如使用叔丁基-(1-(2-氯苯基)-3-羟基-2-氧亚基环己基)氨基甲酸酯的合成的实施例通过如下文的项目4.1解释的鲁伯特姆氧化来实现。
[0791]
此外,在本发明的上下文中,更优选的是,如果进行2-氨基基团或任选地6-羟基基团的修饰,特别地烷基化,其在要点4.2下的2-(2-氯苯基)-2-(二甲基氨基)-6-甲氧基环己酮的合成的实施例中进行解释。
[0792]
4.1.环状2-氨基-1-酮衍生物的6-羟基化的特定实施例
[0793]
向920mg的(2-氧亚基-1-苯基环己基)氨基甲酸叔丁酯(2.85mmol,1.0eq)在15ml的无水thf中的溶液中,在-78℃下在氮气气氛下在7分钟内将3.7ml的二异丙基氨基锂(在thf中的2m溶液,7.40mmol,2.6eq)逐滴加入。将溶液在-78℃下搅拌1小时,然后升温至室温5分钟,然后冷却回到-78℃。在这个温度下,将942μl的三甲基氯硅烷(7.40mmol,2.6eq.)加入以及在-78℃下再搅拌30分钟,以及然后在1小时内升温至室温。
[0794]
将反应通过加入10ml的饱和nh4cl溶液终止,用乙酸乙酯(3x10 ml)萃取水相,将合并的有机相用饱和nacl溶液洗涤,经na2so4干燥,过滤,以及在减压下去除溶剂,因此分离出呈粗产物的淡黄色油。
[0795]
向这种粗产物在15ml的ch2cl2中的溶液中,在-15℃下将539mg的间-氯过苯甲酸(3.13mmol,1.1eq.)加入,并且在这个温度下搅拌1小时。然后将反应混合物升温到室温,用15ml的ch2cl2稀释,以及与10ml的1:1的饱和na2s2o3和饱和nahco3溶液的混合物混合。将水相用ch2cl2(3x10ml)萃取,将合并的有机相用饱和nacl溶液洗涤,经na2so4干燥,过滤并在真空中浓缩。
[0796]
将获得的残余物溶解在15ml的thf中,在-5℃下加入到1.08g的tbaf
·
3h2o(3.42mmol,1.2eq.)在3.42ml的thf中的溶液中,以及在这个温度下搅拌10min。然后加入7ml的饱和nahco3溶液,用乙酸乙酯(3x10ml)萃取水相,将合并的有机相经na2so4干燥,过滤,以及在真空中去除溶剂。通过柱色谱法在硅胶上用乙酸乙酯梯度与环己烷(0
→
60%)纯化残余物。获得523mg的(1-(2-氯苯基)-3-羟基-2-氧亚基环己基)氨基甲酸叔丁酯,呈淡黄色树脂(1.54mmol,36%)。
[0797]
4.2.2-(2-氯苯基)-2-(二甲基氨基)-6-甲氧基环己酮的合成的特定实施例
[0798]
向溶解在4ml的thf中的200mg的(1-(2-氯苯基)-3-羟基-2-氧亚基环己基)氨基甲酸叔丁酯(0.59mmol,1.00eq.)中,在氮气气氛下将684mg的氧化银(i)(2.95mmol,5.00eq.)、403μl的碘甲烷(6.47mmol,11.00eq.)和176mg的粉状活化分子筛加入。将悬浮液加热至40℃持续6小时。
[0799]
然后使用注射器式过滤器去除固体以及在真空中浓缩滤液。通过柱色谱法在硅胶上(0
→
15%乙酸乙酯与环己烷)纯化残余物。获得180mg的(1-(2-氯苯基)-3-甲氧基-2-氧亚基环己基)氨基甲酸叔丁酯,呈无色固体(0.51mmol,86%)。
[0800]
接下来,将37mg的(1-(2-氯苯基)-3-甲氧基-2-氧亚基环己基)氨基甲酸叔丁酯(0.11mmol,1.00eq.)溶解在1ml的ch2cl2中以及将81μl的三氟乙酸(1.10mmol,10.00eq.)加
入。在室温下将溶液搅拌4.5小时以及然后减压去除溶剂。
[0801]
将残余物溶解在1ml的水中以及与1ml的1:1的饱和nahco3和饱和k2co3溶液的混合物混合。将水相用乙酸乙酯(3x3 ml)萃取,将合并的有机相经na2so4干燥,过滤并在真空中将溶剂去除。获得29mg的2-氨基-2-(2-氯苯基)-6-甲氧基环己酮,呈浑浊淡黄色油(0.11mmol,99%)。
[0802]
在单独设置中,向47mg的2-氨基-2-(2-氯苯基)-6-甲氧基环己酮(0.19mmol,1.00eq.)和139μl的37%甲醛水性溶液(1.85mmol,10.00eq.)的溶液中,加入溶解在1ml的乙腈中的37mg的氰基硼氢化钠(0.59mmol,3.20eq.)以及在室温下搅拌15min。将ph定期检查以及使用浓缩的乙酸调节到ph7。在室温下将反应溶液搅拌1小时。
[0803]
然后将溶剂在真空中去除以及将残余物溶于5ml的饱和nahco3溶液中。将水相用ch2cl2(3x3 ml)萃取,将合并的有机相经na2so4干燥,过滤并在真空中将滤液从溶剂中释放。通过柱色谱法在硅胶上(0
→
60%乙酸乙酯与环己烷)纯化残余物。获得36mg的2-(2-氯苯基)-2-(二甲基氨基)-6-甲氧基环己酮(0.13mmol,69%)。
[0804]
4.3.由(2-氧亚基-1-苯基环己基)氨基甲酸叔丁酯合成rac-(2r,6r)-2-氨基-6-羟基-2-苯基环己酮的特定实施例
[0805]
在氮气气氛下,将1.4ml的三乙基硅烷(8.76mmol,29.21eq.)与12滴的铂(0)-1,3-二乙烯基-1,1,3,3-四甲基二硅氧烷络合物在二甲苯中的2%溶液混合以及在室温下搅拌15分钟。然后将87mg(0.30mmol,1.00eq.)的(2-氧亚基-1-苯基环己基)氨基甲酸叔丁酯在2.8ml的thf中的溶液逐滴加入以及在室温下搅拌21小时。最终淡黄色溶液随着反应的进行变为绿棕色。将反应溶液然后在减压下浓缩并且通过柱色谱法在硅胶上用乙酸乙酯梯度与环己烷(0
→
10%)进行纯化。分离出呈灰棕色油的112mg的相应的硅烯醇醚(0.28mmol,93%)以及直接进行进一步的反应。
[0806]
为了这个目的,在氮气气氛下将112mg(0.28mmol,1.00eq.)的硅烯醇醚溶解在2.52ml的二氯甲烷中以及冷却至-15℃。在这个温度下,加入90mg的间-氯过苯甲酸(5.52mmol,1.90eq.)以及将反应混合物搅拌2小时。然后加入2ml的饱和na2so3溶液以及用二氯甲烷(3x5 ml)萃取水相。将合并的有机相用饱和nacl溶液洗涤,经na2so4干燥以及在减压下去除溶剂。
[0807]
将残余物溶解在2.75ml的thf中以及将550μl的四丁基氟化铵在thf中的1m溶液(0.55mmol,2.00eq.)加入。将反应混合物在室温下搅拌2h以及然后将2ml的饱和nahco3溶液和2ml的二氯甲烷加入。将有机相分离以及将水相用二氯甲烷(2x2 ml)萃取两次以上。将合并的有机相经na2so4干燥,过滤并在减压下去除溶剂。通过柱色谱法在硅胶上(0
→
20%乙酸乙酯与环己烷)纯化残余物。分离42mg的((1r,3r)-3-羟基-2-氧亚基-1-苯基环己基)氨基甲酸rac-叔丁酯(0.14mmol,50%),呈无色油。
[0808]
将获得的42mg(0.14mmol,1.00eq.)的((1r,3r)-3-羟基-2-氧亚基-1-苯基环己基)氨基甲酸rac-叔丁酯溶解在1.4ml的二氯甲烷中以及将106μl(1.40mmol,10.00eq.)三氟乙酸加入到溶液中。在室温下搅拌6小时之后,将溶剂使用旋转蒸发器去除以及使残余物与3ml的1:1的饱和na2co3和饱和nahco3溶液的混合物反应。将水相用二氯甲烷(3x5 ml)萃取,将合并的有机相经na2so4干燥,过滤并在减压下将溶剂去除。获得28mg的rac-(2r,6r)-2-氨基-6-羟基-2-苯基环己酮(0.14mmol,98%),呈黄色固体。
[0809]
4.4.制备的经进一步修饰的环状2-氨基-1-酮衍生物的概述和表征
[0810]
下表给出了根据本发明制备的经修饰的环状2-氨基-1-酮衍生物以及在每种情况下获得的产率的概述。
[0811]
[0812]
[0813]
[0814]
[0815][0816]
5.选择的环状2-氨基-1-酮衍生物对具有先前降低的神经元或突触可塑性的神经元的神经系统变性效果的展示
[0817]
为了研究本发明的环状2-氨基-1-酮衍生物的潜在神经系统变性效果,对小鼠胚胎的神经元进行孵育实验。
[0818]
在图1中示出的本发明的环状2-氨基-1-酮衍生物用于这个目的。
[0819]
在详细讨论孵育实验的结果之前,基本孵育测定的设置描述如下。
[0820]
5.1.孵育测定设计和实验程序
[0821]
海马神经元培养物的制备
[0822]
将妊娠小鼠通过颈椎脱位(cervical dislocation)处死以及收集e17.5胚胎。
[0823]
为了保存胚胎的海马,将胚胎杀头以及使用精细的弯曲钳(forceps)打开颅骨。将皮肤和颅侧骨去除以及将整个脑从颅骨中解剖出。在立体显微镜下将脑转移到6cm2的细胞培养皿上,细胞培养皿装填有约5ml的hbss/hepes缓冲液(0.7%hepes)。
[0824]
使用两个精细钳将脑膜从脑上去除以及将海马解剖出并且转移到包含约1ml的hbss/hepes缓冲液的eppendorf管中。然后将分离的海马转移到15ml管以及将缓冲液去除。加入胰蛋白酶(0.05%溶液)(5ml)以及将组织在37℃下在洗浴(wash bath)中孵育15分钟。孵育后,再次将胰蛋白酶溶液去除以及将海马使用hbss/hepes缓冲液洗涤三次并且在3ml的hbss/hepes中使用玻璃巴斯德吸管(pasteur pipette)上下移液进行解离。
[0825]
使用大约原始直径一半的经火抛光的巴斯德吸管通过上下移液进一步分离神经元。使用血细胞计数器(hemocytometer)确定细胞密度,以及以密度为170,000个细胞/6cm2将神经元铺板到在mem(伊格尔极限必需培养基(minimum essential medium eagle):1x mem,0.5%葡萄糖,0.2%nahco3,2mm l-谷氨酰胺,1x mem必需氨基酸,2x mem非必需氨基酸)连同10%fcs(mem-fcs)中的8个经酸处理和聚赖氨酸涂覆的盖玻片上。
[0826]
铺板后24小时,将包含神经元的盖玻片转移到包含大约30-50%汇合的星形胶质细胞的6cm2皿上,所述皿已经提前24小时使用n2培养基(1x mem,0.5%葡萄糖,0.2%丙酮酸,1x neuropan 2(pan biotech,p07-11010))进行平衡。
[0827]
最终,在37℃的5%co2培养箱中培养神经元,其中每周一次更换n2培养基。
[0828]
星形胶质细胞培养
[0829]
将来自e18.5胚胎的星形胶质细胞用作海马神经元的共培养物。
[0830]
如对于培养海马神经元所描述的解剖脑。从胚胎中获得皮质而不是海马。首先以与海马相同的方式处理皮质。
[0831]
然后,将解离的细胞悬浮液再悬浮于10ml的mem-fcs中以及铺板在75cm2的烧瓶上(大约3个脑/烧瓶)。在烧瓶中的星形胶质细胞成长到100%汇合以及分到3个新烧瓶中。为了这个目的,在将细胞悬浮液均等地分到3个新烧瓶中之前,将细胞使用hbss/hepes缓冲液洗涤以及使用胰蛋白酶溶液(0.05%胰蛋白酶)在37℃下孵育5-10min。
[0832]
将包含90-100%汇合的星形胶质细胞的75cm2烧瓶铺开在30x6 cm2的皿上作为海马神经元的共培养物。然后将细胞培养在37℃的5%co2培养箱中,其中每周两次更换培养基。
[0833]
药品刺激
[0834]
在21天的培养之后,将细胞暴露于对照(水)、作为参照的盐酸氯胺酮(ket-h),以及以在水中浓度为0.5μm、1μm和2μm的呈它们各自的盐酸盐的形式的本发明的环状2-氨基-1-酮衍生物(hw-74、-182、-212、-252、-273),以及用它们孵育48和72小时。随后,将细胞用hbss/hepes缓冲液洗涤以及在37℃下在pbs缓冲液中使用4%多聚甲醛固定15分钟。
[0835]
免疫荧光染色
[0836]
在湿度箱中将受刺激的海马神经元染色。为了这个目的,使用tbs/0.2%tritonx-100溶液(tbs-t)对神经元在室温下透化1分钟以及通过在室温下使用氯化铵(50mm)孵育10分钟将自体荧光淬火。通过在4℃下使用tbs-t/10%fcs溶液孵育来阻断非特异性结合位点。
[0837]
将神经元在阻断溶液中在4℃下使用一抗(β-iii-微管蛋白单克隆小鼠igg1(1:500,promega,g7121);小突触小泡蛋白2/vamp2多克隆兔抗血清(1:1000,synaptic systems,104202))进行孵育。这之后是通过在tbs-t中温和地浸渍进行洗涤三次,以及加入一滴pbs。
[0838]
然后将二抗(抗小鼠igg-atto488(1:1000,sigma-aldrich,62197;抗兔igg-atto550(1:1000,sigma-aldrich,43328))加入到阻断溶液中以及在室温下孵育2小时,然后以与前文描述的相同的方式洗涤。
[0839]
为了对细胞核进行染色,使用在pbs中稀释的dna染料dapi(10ng/ml)在室温下将神经元孵育10分钟。最后,使用pbs简单洗涤神经元,然后将神经元转移到包含每个载玻片大约10μl的mowiol包埋剂的载玻片。
[0840]
神经元棘结构的密度分析。
[0841]
使用zeiss axio observer在25x的放大率下拍摄制备的神经元的荧光图像。使用zen软件(zeiss)进行突触扣结的数量的分析。随机选择40μm的近端树突段(每个神经元3个)以及使用预设强度阈值检测用于vamp2染色的信号点。相对于对于对照(未处理的)细胞获得的扣结的数量,表示结果。
[0842]
5.2.孵育测定的结果
[0843]
用根据本发明的上文提及的环状2-氨基-1-酮衍生物的孵育实验结果的概述,可以在图2至4中看到。
[0844]
图2给出了在用根据本发明的化合物以及参照盐酸氯胺酮(ket-h)和对照水孵育
制备的神经元后48小时内观察的化合物hw-74、hw-182、hw-212、hw-252和hw-273的神经系统变性效果的概述。
[0845]
这里绘制的是针对不同浓度(0.25μm、0.5μm、1μm、2μm、5μm和10μm)的本发明的环状2-氨基-1-酮衍生物和ket-h分别相对于对照的突触前扣结的数量。
[0846]
对于几乎所有的化合物,除了hw-182,观察到在突触前扣结的形成中浓度依赖性增加。此处,与对照相比,衍生物hw-212在突触前扣结的数量上引起了中等增加。特别地对于化合物hw-252和hw-273,可以观察到产生的突触前扣结显著增加,其特别地与高神经系统变性潜能相关。在此上下文中,特别值得注意的是衍生物hw-252和hw-273的功效明显超过了母体化合物ket-h的功效,使得这里可以认为活性特性和分子结构的相对的优势和显著的改善。
[0847]
图3示出了在用前文提及的根据本发明的化合物以及参照盐酸氯胺酮(ket-h)和对照水孵育制备的神经元72小时后孵育测定的结果。
[0848]
与图2中示出的结果相比,可以看出根据本发明的化合物,特别地衍生物hw-252和hw-273从长远来看,即使在低剂量下,具有显著的神经系统变性效果,以及有效地刺激新的突触前扣结的形成。
[0849]
此外,结果确认根据本发明的化合物基本上长期有效,因为它们能够以持续的方式诱导突触前扣结的新形成。
[0850]
此外,图4示出了用化合物hw-94、hw-157、hw-159、hw-195、hw-216、hw-224、hw-229、hw-245、hw-247和hw-283,对比使用盐酸氯胺酮(ket-h)作为参照和水作为对照进行48小时孵育之后的孵育测定的结果。上文提及的化合物已经以浓度0.5μm、1μm、2μm和5μm使用。发现甲氧基取代的衍生物hw-94、hw-195和hw-283在孵育测定中相对最有效,诱导产生的突触前扣结显著增加。总之,还已经观察到本发明的化合物的效果在48小时的时间段内是特别显著的并且超过该时间持续并且逐渐降低。
[0851]
最后,图5示出了用衍生物hw-273以三种不同的浓度以及在72小时的时间段内孵育后制备的神经元的荧光显微镜图像。
[0852]
这些图像再次清晰地展示本发明的化合物对神经元的再生的浓度依赖性积极效果。这里特别地值得注意的是,突触前扣结沿着神经元的神经突的强有力地明显的构型以及,尤其是,神经元对加入hw-273响应的加强的生长。未知或尚没有观察到,特别地对于相对的或母体化合物ket-h和羟基去甲氯胺酮的可比较效果。因此,根据本发明的衍生物实现了先前未知的作用潜能,其特别地还远远超出了基本母体化合物的作用模式。
[0853]
关于根据本发明的最有效的衍生物hw-252和hw273的分子结构,最后可以推断,首先特别地在芳基取代基处的低电子密度对于根据本发明的化合物的作用潜能具有有利效果。同样地,如果引入残基,例如烷基残基,其增加官能团,特别地2-氨基基团和任选地6-羟基基团的亲脂性,这是特别地有利的。
[0854]
在此上下文中,根据本发明的方法现在提供了决定性的优点,在每种情况下提及的有益结构要素都可以在本发明的范围内引入,而没有任何问题或更复杂化并且还可以以多种方式变化。因此,根据本发明的方法明确地允许获得基于环状2-氨基-1-酮的广范围的用于治疗神经退行性疾病的有前景的潜在的新活性物质。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。