1.本发明属于化学合成技术领域,具体涉及一种盐酸贝尼地平中间体的制备方法。
背景技术:
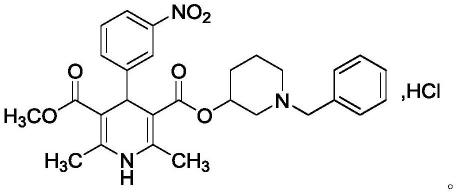
2.盐酸贝尼地平,结构式如下:
[0003][0004]
专利ep106275报道盐酸贝尼地平以1-苄基-3-哌啶醇与2,6-二甲基-4-(3-硝基苯基)-1,4-二氢-3,5-吡啶二甲酸单甲酯为起始原料,在氯化亚砜的条件下,经酯化反应制得盐酸贝尼地平。
[0005][0006]
文献chem.pharm.bull.42(8)1579-1589报道了一种2,6-二甲基-4-(3-硝基苯基)-1,4-二氢-3,5-吡啶二甲酸单甲酯制备方法,该方法以间硝基苯甲醛与乙酰乙酸甲酯为原料,经克脑文格尔缩合制得2-[(3-硝基苯基)亚甲基]-3-氧代丁酸甲酯,再与3-氨基-2-丁烯酸-2-氰基乙基酯(式i化合物)反应后在硫化钠或四丁基氟化铵的条件下水解制得,该方法制得2,6-二甲基-4-(3-硝基苯基)-1,4-二氢-3,5-吡啶二甲酸单甲酯水解收率高,纯度高,二酸杂质少,适合工业化生产。
[0007][0008]
目前,报道的中间体式ⅰ化合物的合成方法主要有:
[0009]
(1)武汉百科药物开发有限公司申请专利cn200810246353以3-羟基丙腈与双乙烯酮为原料反应制得式ⅳ化合物,式ⅳ化合物再经一步氨化即可制得式ⅰ化合物。
[0010][0011]
该方法制备式ⅳ化合物收率只有54%,并且所用原料双乙烯酮性质活泼,具有强烈的刺激作用和催泪性,需低温保存与运输;且反应过程中急剧放热,工业化生产时有一定的安全隐患。
[0012]
(2)威海迪素制药有限公司申请cn201310692163以3-羟基丙腈与乙酰乙酸甲酯为原料,经酯交换反应制得式ⅳ化合物,制得式ⅳ化合物再经氨化反应制得式ⅰ化合物。
[0013][0014]
此方法原料易得,但在现有报道中此方法的后处理麻烦,在制备式ⅳ化合物需要甲苯做溶剂,后处理需要经过水洗,旋干等操作,由于甲苯沸点110.6℃,在生产上进行蒸馏操作十分耗时,耗能;且制备式ⅰ化合物时反应时间需要18小时,因此,该方法不适合工业化生产。
技术实现要素:
[0015]
有鉴于此,本发明的目的之一在于提供一种无溶剂制备ⅳ化合物的方法,该方法制备过程中无需添加任何其他溶剂,适合工业化生产。
[0016]
为实现上述目的,本发明采用以下技术方案:
[0017]
一种无溶剂制备ⅳ化合物的方法,以式ⅱ化合物和式ⅲ化合物为原料,不添加溶剂,进行酯交换反应,得反应产物1;所述反应产物1含有ⅳ化合物;
[0018][0019]
进一步,式ⅱ化合物为3-羟基丙腈,式ⅲ化合物为乙酰乙酸甲酯,式ⅳ化合物为3-氧代丁酸-2-氰基乙基酯。
[0020]
进一步,所述酯交换反应过程中产生副产物甲醇,所述副产物甲醇的去除方式为常压蒸馏、减压蒸馏、体系通入空气、体系通入氮气中的任一种或多种。
[0021]
更进一步,去除副产物甲醇的方法优选为反应过程中体系通入氮气。
[0022]
进一步,所述式ⅱ化合物和所述式ⅲ化合物的摩尔比为1:1.5~3。
[0023]
更进一步,式ⅱ化合物和式ⅲ化合物的摩尔比优选为1:1.5。
[0024]
进一步,制备过程中添加催化剂三氟化硼乙醚、硼酸和/或溴代丁二酰亚胺,或不加催化剂。
[0025]
更进一步,所述催化剂优选为硼酸。
[0026]
进一步,所述式ⅱ化合物和所述硼酸的摩尔比为1:0.005~0.1,优选为1:0.01。
[0027]
进一步,所述酯交换反应的反应温度为100℃~120℃,优选为105℃~115℃。
[0028]
进一步,酯交换反应完成后,不进行其他操作,直接降温至室温。
[0029]
本发明的目的之二在于提供一种用反应产物1制备式ⅰ化合物的方法,所述式ⅰ化合物为3-氨基-2-丁烯酸-2-氰基乙基酯,是盐酸贝尼地平的一个关键中间体,同时也是地平类药物的关键中间体,目前的文献报道中,其合成操作方法较为复杂耗时,不适宜工业化生产,本发明填补了空白。
[0030]
为实现上述目的,本发明采用以下技术方案:
[0031]
一种用反应产物1制备式ⅰ化合物的方法,所述式ⅳ化合物和酯交换反应剩余的式ⅲ化合物发生氨化反应,得反应产物2;所述反应产物2含有式ⅰ化合物;
[0032][0033]
进一步,所述氨化反应的反应温度为10℃~50℃,优选为20℃~30℃。
[0034]
进一步,反应溶剂包括甲醇、乙醇、异丙醇、四氢呋喃、水中的任一种或多种。
[0035]
更进一步,反应溶剂优选为甲醇/水(体积比)=1.5:1。
[0036]
进一步,所述反应溶剂与所述式ⅱ化合物的体积质量比为8~12:1。
[0037]
更进一步,所述反应溶剂与式ⅱ化合物的体积质量比优选为10:1。
[0038]
进一步,氨化反应试剂为乙酸铵、氯化铵、碳酸铵、氨基甲酸铵中的任一种或多种。
[0039]
更进一步,所述氨化反应试剂优选为乙酸铵。
[0040]
进一步,所述氨化反应试剂与所述式ⅱ化合物的摩尔比为3~9:1。
[0041]
更进一步,所述氨化反应试剂与式ⅱ化合物的摩尔比优选为4:1。
[0042]
进一步,所述反应产物2中还含有式
ⅴ
化合物,所述反应产物2经精制和干燥除去式
ⅴ
化合物,得到式ⅰ化合物;
[0043][0044]
进一步,式
ⅴ
化合物为3-氨基丁-2-烯酸甲酯。
[0045]
进一步,精制溶剂为甲醇、乙醇、异丙醇、四氢呋喃、水中的任一种或多种。
[0046]
更进一步,所述精制溶剂优选为水。
[0047]
进一步,所述精制溶剂与所述式ⅱ化合物体积质量比为8~15:1。
[0048]
更进一步,所述精制溶剂与所述式ⅱ化合物体积质量比优选为12:1。
[0049]
本发明的目的之三在于提供一种制备所述式ⅰ化合物的生产系统。
[0050]
为实现上述目的,本发明采用以下技术方案:
[0051]
一种制备所述式ⅰ化合物的生产系统,所述生产系统包含生产单元1、副产物处理单元、生产单元2和精制单元,不包含溶剂处理单元;所述生产单元1、副产物处理单元、生产单元2和精制单元依次连接;所述式ⅱ化合物和所述式ⅲ化合物在生产单元1中进行酯交换反应,并在副产物处理单元中除去副产物甲醇,得所述反应产物1;所述反应产物1中的式ⅳ化合物和反应剩余的式ⅲ化合物在生产单元2中进行氨化反应,得反应产物2;所述反应产
物2在精制单元进行精制和干燥,得到式ⅰ化合物。
[0052]
反应过程如下:
[0053][0054]
本发明的有益效果在于:
[0055]
1.本发明提供的盐酸贝尼地平中间体(3-氨基-2-丁烯酸-2-氰基乙基酯)的制备方法,以乙酰乙酸甲酯和3-羟基丙腈为原料,通过酯交换反应得到3-氧代丁酸-2-氰基乙基酯(式ⅳ化合物),反应完成后,直接降温,反应体系进行氨化反应得式ⅰ化合物和式
ⅴ
化合物的混合物,最后经精制和干燥得到纯的3-氨基-2-丁烯酸-2-氰基乙基酯(式ⅰ化合物),该方法具有收率高,成本低,操作简单,适合工业化生产等特点。
[0056]
2.本发明公开的盐酸贝尼地平中间体(式ⅰ化合物:-氨基-2-丁烯酸-2-氰基乙基酯)的制备方法,对于盐酸贝尼地平工业化生产具有重大意义,它可以用于大批量生产盐酸贝尼地平中间体,节约成本,提高效率。
附图说明
[0057]
图1为实施例1的色谱图;
[0058]
图2为实施例2的色谱图;
[0059]
图3为实施例3的色谱图;
[0060]
图4为实施例4的色谱图;
[0061]
图5为实施例5的色谱图。
具体实施方式
[0062]
下面将结合具体的实施例对本发明的技术方案进行更进一步地清楚、完整地描述。显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部实施例。因此,基于本发明中的实施例,本领域技术人员在没有付出创造性劳动前提下所获得的其他所有实施例都属于本发明的保护范围。
[0063]
以下实施例中,eq表示当量,与主原料的比例。
[0064]
实施例1.3-氨基-2-丁烯酸-2-氰基乙基酯(式ⅰ化合物)的制备
[0065]
1.酯交换反应制备ⅳ化合物:在三口瓶中,加入3-羟基丙腈50g(1.0eq),乙酰乙酸甲酯163.5g(2.0eq),硼酸0.4g(0.01eq),搅拌升温至100℃~105℃,抽真空,减压蒸馏,反应10小时后,取样中控,直至反应达到平衡,3-羟基丙腈含量基本不再变化,降温至室温。
[0066]
2.氨化反应制备式ⅰ化合物:三口瓶中加入水250ml,乙酸铵325.8g(6.0eq),步骤1制得的反应液和乙醇250ml,搅拌,于10℃~15℃反应2小时,降温至0℃~5℃,析晶1小时,抽滤,并用水淋洗滤饼。
[0067]
3.精制、干燥:三口瓶中加入水500ml,步骤2制得的滤饼,室温搅拌1小时,抽滤,并用水淋洗滤饼,减压干燥,得到3-氨基-2-丁烯酸-2-氰基乙基酯(式ⅰ化合物)67.3g,总收率62.0%,hplc检测式ⅰ化合物的纯度为99.76%(式
ⅴ
化合物0.24%),液相色谱积分结果如表1所示,色谱图如图1所示。
[0068]
表1.实施例1的液相色谱积分结果
[0069][0070]
实施例2.3-氨基-2-丁烯酸-2-氰基乙基酯(式ⅰ化合物)的制备
[0071]
1.酯交换反应制备ⅳ化合物:三口瓶中,加入3-羟基丙腈50g(1.0eq),乙酰乙酸甲酯204.4g(2.5eq),三氟化硼乙醚1.0g(0.01eq),搅拌升温至100℃~105℃,氮气通入液面下,常压蒸馏,反应10小时后,取样中控,直至反应达到平衡,3-羟基丙腈含量基本不再变化,降温至室温。
[0072]
2.氨化反应制备式ⅰ化合物:三口瓶中加入乙醇500ml,氯化铵188.4g(5.0eq),步骤1制得的反应液,于20℃~25℃反应2小时,降温至0℃~5℃,析晶1小时,抽滤,并用水淋洗滤饼。
[0073]
3.精制、干燥:三口瓶中加入水600ml,步骤2制得的滤饼,室温搅拌1小时,抽滤,并用水淋洗滤饼,减压干燥,得到3-氨基-2-丁烯酸-2-氰基乙基酯63.6g,总收率58.6%,hplc检测式ⅰ化合物纯度为99.65%(式
ⅴ
化合物0.35%),液相色谱积分结果如表2所示,色谱图如图2所示。
[0074]
表2.实施例2的液相色谱积分结果
[0075][0076]
实施例3.3-氨基-2-丁烯酸-2-氰基乙基酯(式ⅰ化合物)的制备
[0077]
1.酯交换反应制备ⅳ化合物:三口瓶中加入3-羟基丙腈100g(1.0eq),乙酰乙酸甲酯245.3g(1.5eq),搅拌升温至105℃~110℃,常压蒸馏,反应10小时后,取样中控,直至反应达到平衡,3-羟基丙腈含量基本不再变化,降温至室温。
[0078]
2.氨化反应制备式ⅰ化合物:三口瓶中加入水500ml,碳酸铵676.8g(5.0eq),步骤1制得的反应液和异丙醇500ml,于20℃~30℃反应2小时,降温至0℃~5℃,析晶1小时,抽滤,并用水淋洗滤饼。
[0079]
3.精制、干燥:三口瓶中加入水600ml和600ml甲醇,搅拌加入步骤2离心所得固体,室温搅拌1小时,抽滤,并用水淋洗滤饼,减压干燥,得到3-氨基-2-丁烯酸-2-氰基乙基酯
133.5g,总收率61.5%,hplc检测式ⅰ化合物纯度为99.75%(式
ⅴ
化合物0.25%),液相色谱积分结果如表3所示,色谱图如图3所示。
[0080]
表3.实施例3的液相色谱积分结果
[0081][0082][0083]
实施例4.3-氨基-2-丁烯酸-2-氰基乙基酯(式ⅰ化合物)的制备
[0084]
1.酯交换反应制备ⅳ化合物:30l反应釜中,加入3-羟基丙腈5.0kg(1.0eq),乙酰乙酸甲酯24.5kg(3.0eq),搅拌升温至110℃~115℃,通入氮气,反应10小时后,取样中控,直至反应达到平衡,3-羟基丙腈含量基本不再变化,降温至室温。
[0085]
2.氨化反应制备式ⅰ化合物:80l反应釜中加入纯化水25.0kg,搅拌加入乙酸铵27.11kg(5.0eq),溶清后,加入步骤1制得的反应液和甲醇19.75kg,控制物料温度25℃~30℃,反应2小时,降温至0℃~5℃,析晶1小时,离心,并用水淋洗滤饼。
[0086]
3.精制、干燥:50l反应釜中加入水25.0kg,搅拌加入步骤2离心所得固体,室温搅拌1小时,离心,并用水淋洗滤饼,减压干燥,得到3-氨基-2-丁烯酸-2-氰基乙基酯6.32kg,总收率58.2%,hplc检测式ⅰ化合物纯度为99.37%(式
ⅴ
化合物0.62%),液相色谱积分结果如表4所示,色谱图如图4所示。
[0087]
表4.实施例4的液相色谱积分结果
[0088][0089]
实施例5.3-氨基-2-丁烯酸-2-氰基乙基酯(式ⅰ化合物)的制备
[0090]
1.酯交换反应制备ⅳ化合物:30l反应釜中,加入3-羟基丙腈5.0kg(1.0eq),乙酰乙酸甲酯12.25kg(1.5eq),硼酸43.5g,搅拌升温至105℃~115℃,通入氮气,常压蒸馏,反应10小时后,取样中控,直至反应达到平衡,3-羟基丙腈含量基本不再变化,降温至室温。
[0091]
2.氨化反应制备式ⅰ化合物:80l反应釜中加入纯化水15.0kg,搅拌加入乙酸铵21.7kg(4.0eq),溶清后,加入步骤1制得的反应液和甲醇17.8kg,控制物料温度20℃~25℃,反应2小时,降温至0℃~5℃,析晶1小时,离心,并用水淋洗滤饼。
[0092]
3.精制、干燥:50l反应釜中加入水25.0kg,搅拌加入步骤2离心所得固体,室温搅拌1小时,离心,并用水淋洗滤饼,减压干燥,得到3-氨基-2-丁烯酸-2-氰基乙基酯6.62kg,总收率61.0%,hplc检测式ⅰ化合物纯度为99.55%(式
ⅴ
化合物0.45%),液相色谱积分结果如表5所示,色谱图如图5所示。
[0093]
表5.实施例5的液相色谱积分结果
[0094]
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
