1.本发明涉及有机合成领域。更具体地说,本发明涉及一种胆固醇硒氰酸酯类化合物及其制备方法与应用。
背景技术:
2.硒元素作为生物体的必需微量元素,在人类的生长及发育过程中发挥着极其重要的生理作用。有机硒化合物在抗氧化、抗炎、抗肿瘤等方面展现出卓越的生物活性,对有机硒化合物的开发应用研究是目前化学、生命科学、临床医学等研究领域的一个热点(organoselenium small molecules and chromium(iii)complexes for intervention in chronic low-grade inflammation and type 2 diabetes[j].current topics in medicinal chemistry,2016:16(8):823-34)。硒氰酸酯化合物作为一类有机硒化合物,通常显示出较好生物活性(cui j g,pang l p,wei m z,huang y m,et al.synthesis and antiproliferative activity of 17-[1
′
,2
′
,3
′
]-selenadiazolylpregnenolone compounds,steroids,2018,140,151-158;huang y m,peng z n,wei m z,et al.synthesis and antiproliferative evaluation of some novel estradiol selenocyanates[j].steroids,2022,181:108992),同时也是合成例如硒醚,二硒醚,硒脲等有机硒化合物的中间体,而这些中间体具有潜在的药用价值。
[0003]
胆固醇是哺乳动物细胞膜的重要脂质成分,用以维持细胞膜的完整性和流动性,也是所有类固醇激素和维生素d类似物的主干,它对生命的生长发育起着重要的作用。近年来针对胆固醇代谢的新分子和新策略的筛选已成为癌症研究领域的热点问题,一些胆固醇代谢分子,如soat1、sqle和npc1,已经发展成为治疗癌症的靶点药物,但关于胆固醇硒氰酸酯类化合物在抗肿瘤药物方面的研究却很少,而且现有技术的药物通常只对一两种肿瘤细胞有抑制作用。因此,寻求一种具有更加广泛抑制作用的药物是非常有必要的。
技术实现要素:
[0004]
本发明的一个目的是解决至少上述问题,并提供至少后面将说明的优点。
[0005]
为了实现根据本发明的这些目的和其它优点,提供了一种胆固醇硒氰酸酯类化合物,具有通式i所示结构:
[0006][0007]
r1为硒氰基乙酰基、硒氰基丁酰基、硒氰基戊酰基、对硒氰基甲基苯甲酰基、间硒氰基甲基苯甲酰基、硒氰基庚酰基、硒氰基十一酰基、硒氰基戊酸酯基中的任意一种。
[0008]
提供一种胆固醇硒氰酸酯类化合物的制备方法,包括以下步骤:
[0009]
步骤一、将胆固醇用二氯甲烷和甲醇溶解,于低于-78℃条件下通入臭氧,并搅拌反应,经薄层层析技术监测胆固醇反应完全后,停止通入臭氧,加入二甲硫醚,继续搅拌直至反应结束,将反应结束后的液体,减压蒸馏除去溶剂,得到透明油状的中间产物1;
[0010]
步骤二、将中间产物1用苯溶解,加入中性氧化铝,于室温条件下搅拌直至反应结束,抽滤除去中性氧化铝,用二氯甲烷冲洗滤膜上的中性氧化铝得到滤液,将滤液减压抽滤,得到油状物,将油状物经柱层析分离得到中间产物2;
[0011]
步骤三、将中间产物2用无水乙醇搅拌溶解,加热至70~80℃条件下,在加热过程中加入三水乙酸钠并搅拌,加入盐酸羟胺,然后回流搅拌,直至反应结束,将反应结束后的液体经萃取得到有机相,有机相经洗涤、干燥、浓缩后得到粗产物ⅰ,粗产物ⅰ经柱层析分离得到中间产物3;
[0012]
步骤四、将中间产物3用无水乙醇加热搅拌溶解,依次加入氰基硼氢化钠、五氯化钼和一水合硫酸氢钠,于室温条件下继续搅拌,直至反应结束,反应结束后的液体经萃取得到有机相,有机相经洗涤、干燥、浓缩后得到粗产物ⅱ,粗产物ⅱ经柱层析分离得到中间产物4;
[0013]
步骤五、将中间产物4用二氯甲烷搅拌溶解,加入三乙胺,于0℃条件下并搅拌15~20min,滴加酰氯化合物,继续搅拌30~40min,于室温条件下搅拌,直至反应结束,反应结束后的液体经萃取得到有机相,有机相经洗涤、干燥、浓缩后得到粗产物ⅲ,粗产物ⅲ经柱层析分离得到中间产物5,其中,酰氯化合物为氯乙酰氯、4-氯丁酰氯、5-氯代戊酰氯、4-氯甲基苯甲酰氯、3-氯甲基苯甲酰氯中的任意一种;
[0014]
其中,当酰氯化合物为氯乙酰氯时,得到中间产物5a:
[0015][0016]
当酰氯化合物为4-氯丁酰氯时,得到中间产物5b:
[0017][0018]
当酰氯化合物为5-氯代戊酰氯时,得到中间产物5c:
[0019][0020]
当酰氯化合物为4-氯甲基苯甲酰氯时,得到中间产物5d:
[0021][0022]
当酰氯化合物为3-氯甲基苯甲酰氯时,得到中间产物5e:
[0023][0024]
步骤六、将中间产物5用n,n-二甲基甲酰胺搅拌溶解,加入硒氰酸钾溶液,通入氩气,并用锡纸包裹,于70~80℃条件下搅拌,直至反应结束,反应结束后的液体经萃取得到有机相,有机相经洗涤、干燥、浓缩后得到粗产物ⅳ,粗产物ⅳ经柱层析分离得到具有通式i结构的胆固醇硒氰酸酯化合物6a~6e;
[0025]
其中,当步骤五中的酰氯化合物为氯乙酰氯时,得到胆固醇硒氰酸酯化合物6a:
[0026][0027]
当步骤五中的酰氯化合物为4-氯丁酰氯时,得到胆固醇硒氰酸酯化合物6b:
[0028][0029]
当步骤五中的酰氯化合物为5-氯代戊酰氯时,得到胆固醇硒氰酸酯化合物6c:
[0030][0031]
当步骤五中的酰氯化合物为4-氯甲基苯甲酰氯时,得到胆固醇硒氰酸酯化合物6d:
[0032][0033]
当步骤五中的酰氯化合物为3-氯甲基苯甲酰氯时,得到胆固醇硒氰酸酯化合物6e:
[0034][0035]
优选的是,还包括以下步骤:
[0036]
步骤五、在氩气保护条件下,往中间产物4中依次加入溴酸化合物、六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷、4-二甲氨基吡啶、三乙胺以及二氯甲烷,于室温条件下搅拌,直至反应结束,反应结束后的液体经萃取得到有机相,有机相经洗涤、干燥、浓缩后得到粗产物
ⅴ
,粗产物
ⅴ
经柱层析分离得到中间产物6,其中,所述溴酸化合物为7-溴庚酸、溴代十一酸、4-(2-溴乙氧基)苯甲酸中的任意一种;
[0037]
步骤六、将中间产物6用n,n-二甲基甲酰胺搅拌溶解后,加入硒氰酸钾溶液,通入氩气,并用锡纸包裹,于70~80℃条件下搅拌,直至反应结束,反应结束后的液体经萃取得到有机相,有机相经洗涤、干燥、浓缩后得到粗产物ⅵ,粗产物ⅵ经柱层析分离得到具有通式i结构的胆固醇硒氰酸酯化合物6f~6h;
[0038]
其中,当步骤五中的溴酸化合物为7-溴庚酸时,得到胆固醇硒氰酸酯化合物6f:
[0039][0040]
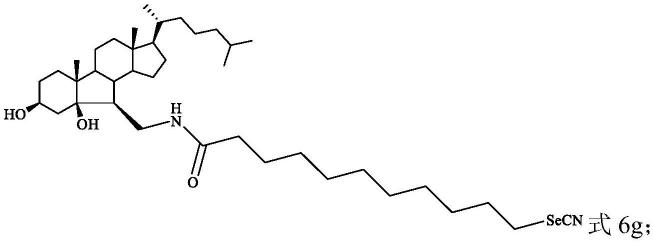
当步骤五中的溴酸化合物为溴代十一酸时,得到胆固醇硒氰酸酯化合物6g:
[0041][0042]
当步骤五中的溴酸化合物为4-(2-溴乙氧基)苯甲酸时,得到胆固醇硒氰酸酯化合
物6h:
[0043][0044]
优选的是,萃取操作用到的溶剂为乙酸乙酯、甲醇、二氯甲烷、四氯化碳中的任意一种。
[0045]
优选的是,柱层析分离操作用的溶剂为乙酸乙酯与石油醚按体积比为1:1~15混合得到。
[0046]
提供一种胆固醇硒氰酸酯类化合物在制备抗肿瘤药物中的应用。
[0047]
本发明至少包括以下有益效果:
[0048]
第一、本发明提供了一种胆固醇硒氰酸酯类化合物及其制备方法,所述胆固醇硒氰酸酯类化合物结构新颖,且其制备方法收率高、底物适用性广。
[0049]
第二、本发明提供的胆固醇硒氰酸酯类化合物具有优异的抗肿瘤活性,其中,对所有受试肿瘤细胞均具有明显的抑制作用,ic
50
值均小于9μmol/l,比市售药物阿比特龙对肿瘤细胞的抑制作用更加优异。
[0050]
本发明的其它优点、目标和特征将部分通过下面的说明体现,部分还将通过对本发明的研究和实践而为本领域的技术人员所理解。
具体实施方式
[0051]
下面结合实施例对本发明做进一步的详细说明,以令本领域技术人员参照说明书文字能够据以实施。
[0052]
需要说明的是,下述实施方案中所述实验方法,如无特殊说明,均为常规方法,所述试剂和材料,如无特殊说明,均可从商业途径获得。
[0053]
《实施例1》
[0054]
b-降-3,5-二羟基胆甾-6-硒氰基乙酰胺(6a)的制备:
[0055]
步骤一、将772mg(2mmol)胆固醇用20ml二氯甲烷和5ml甲醇溶解,于-78℃条件下通入臭氧,并搅拌反应,经薄层层析技术监测胆固醇反应完全后,停止通入臭氧,加入2ml二甲硫醚,继续搅拌直至反应结束,将反应结束后的液体,减压蒸馏除去溶剂,得到透明油状的中间产物1:
[0056][0057]
步骤二、将步骤一中收得的中间产物1用20ml苯溶解,加入3g中性氧化铝,于室温条件下搅拌,直至反应结束,抽滤除去中性氧化铝,用二氯甲烷冲洗滤膜上的中性氧化铝得到滤液,将滤液减压抽滤,得到油状物,将油状物采用乙酸乙酯与石油醚体积比为1:5的硅胶柱层析分离得到443mg透明油状物,即中间产物2:
[0058][0059]
产率为53%,经核磁共振和质谱分析确定中间产物2的结构;
[0060]
步骤三、将175mg(0.42mmol)中间产物2用30ml无水乙醇搅拌溶解,加热至80℃条件下,在加热过程中加入115mg(0.84mmol)三水乙酸钠并搅拌,加入59mg(0.84mmol)盐酸羟胺,然后回流搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅰ,粗产物ⅰ采用乙酸乙酯与石油醚体积比为1:3的硅胶柱层析分离得到154mg白色固体,即中间产物3:
[0061][0062]
产率为85%,通过熔点测定实验得到熔点为:145~147℃,经核磁共振和质谱分析确定中间产物3的结构;
[0063]
步骤四、将325mg(0.75mmol)中间产物3用20ml无水乙醇加热搅拌溶解,依次加入187mg(3.00mmol)氰基硼氢化钠、203mg(0.75mmol)五氯化钼和308mg(2.25mmol)一水合硫酸氢钠,于室温条件下继续搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅱ,粗产物ⅱ采用乙酸乙酯与石油醚体积比为1:15的硅胶柱层析分离得到252mg淡黄色固体,即中间产物4:
[0064][0065]
产率为80%,通过熔点测定实验得到熔点为:121~123℃,经核磁共振和质谱分析确定中间产物4的结构;
[0066]
步骤五、将250mg(0.596mmol)中间产物4用15ml二氯甲烷搅拌溶解,加入250μl(1.788mmol)三乙胺,于0℃条件下搅拌15min,滴加60μl(0.76mmol)氯乙酰氯,继续搅拌30min后,于室温条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅲ,粗产物ⅲ采用乙酸乙酯与石油醚体积比为1:4的硅胶柱层析分离得到137mg白色固体,即中间产物5a:
[0067][0068]
产率为46%,通过熔点测定实验得到熔点为:148~150℃,经核磁共振和质谱分析确定中间产物5a的结构;
[0069]
步骤六、将120mg(0.242mmol)中间产物5a用15ml n,n-二甲基甲酰胺搅拌溶解后,加入1.5mol/l 1ml的硒氰酸钾溶液,通入氩气,并用锡纸包裹,于80℃条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅳ,粗产物ⅳ采用乙酸乙酯与石油醚体积比为1:2的硅胶柱层析分离得到92mg白色固体,即b-降-3,5-二羟基胆甾-6-硒氰基乙酰胺(6a):
[0070][0071]
产率为67%,通过熔点测定实验得到熔点为:165~166℃,经核磁共振和质谱分析确定b-降-3,5-二羟基胆甾-6-硒氰基乙酰胺的结构。
[0072]
《实施例2》
[0073]
b-降-3,5-二羟基胆甾-6-硒氰基丁酰胺(6b)的制备:
[0074]
步骤一、将772mg(2mmol)胆固醇用20ml二氯甲烷和5ml甲醇溶解,在-78℃以下通入臭氧,并搅拌反应,经薄层层析技术监测胆固醇反应完全后,停止通入臭氧,加入2ml二甲硫醚,继续搅拌直至反应结束,将反应结束后的液体,减压蒸馏除去溶剂,得到透明油状的中间产物1:
[0075]
[0076]
步骤二、将步骤一中收得的中间产物1用20ml苯溶解,加入3g中性氧化铝,于室温条件下搅拌,直至反应结束,抽滤除去中性氧化铝,用二氯甲烷冲洗滤膜上的中性氧化铝得到滤液,将滤液减压抽滤,得到油状物,将油状物采用乙酸乙酯与石油醚体积比为1:5的硅胶柱层析分离得到443mg透明油状物,即中间产物2:
[0077][0078]
产率为53%,经核磁共振和质谱分析确定中间产物2的结构;
[0079]
步骤三、将175mg(0.42mmol)中间产物2用30ml无水乙醇搅拌溶解,加热至80℃条件下,在加热过程中加入115mg(0.84mmol)三水乙酸钠并搅拌,加入59mg(0.84mmol)盐酸羟胺,然后回流搅拌直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅰ,粗产物ⅰ采用乙酸乙酯与石油醚体积比为1:3的硅胶柱层析分离得到154mg白色固体,即中间产物3:
[0080][0081]
产率为85%,通过熔点测定实验得到熔点为:145~147℃,经核磁共振和质谱分析确定中间产物3的结构;
[0082]
步骤四、将325mg(0.75mmol)中间产物3用20ml无水乙醇加热搅拌溶解后,依次加入187mg(3.00mmol)氰基硼氢化钠、203mg(0.75mmol)五氯化钼和308mg(2.25mmol)一水合硫酸氢钠,于室温条件下继续搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅱ,粗产物ⅱ采用乙酸乙酯与石油醚体积比为1:15的硅胶柱层析分离得到252mg淡黄色固体,即中间产物4:
[0083][0084]
产率为80%,通过熔点测定实验得到熔点为:121~123℃,经核磁共振和质谱分析确定中间产物4的结构;
[0085]
步骤五、将250mg(0.596mmol)中间产物4用15ml二氯甲烷搅拌溶解后,加入250μl(1.788mmol)三乙胺后,于0℃条件下搅拌15min,滴加87μl(0.76mmol)4-氯丁酰氯,继续搅拌30min后于室温条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物
ⅲ
,粗产物ⅲ采用乙酸乙酯与石油醚体积比为1:4的硅胶柱层析分离得到173mg白色固体,即中间产物5b:
[0086][0087]
产率为55%,通过熔点测定实验得到熔点为:149~151℃,经核磁共振和质谱分析确定中间产物5b的结构;
[0088]
步骤六、将136mg(0.242mmol)中间产物5b用15ml n,n-二甲基甲酰胺搅拌溶解后,加入1.5mol/l 1ml的硒氰酸钾溶液,通入氩气,并用锡纸包裹,于80℃条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅳ,粗产物ⅳ采用乙酸乙酯与石油醚体积比为1:2的硅胶柱层析分离得到100mg白色固体,即b-降-3,5-二羟基胆甾-6-硒氰基丁酰胺(6b):
[0089][0090]
产率为64%,通过熔点测定实验得到熔点为:145~146℃,经核磁共振和质谱分析确定b-降-3,5-二羟基胆甾-6-硒氰基丁酰胺的结构。
[0091]
《实施例3》
[0092]
b-降-3,5-二羟基胆甾-6-硒氰基戊酰胺(6c)的制备:
[0093]
步骤一、将772mg(2mmol)胆固醇用20ml二氯甲烷和5ml甲醇溶解,在-78℃以下通入臭氧,并搅拌反应,经薄层层析技术监测胆固醇反应完全后,停止通入臭氧,加入2ml二甲硫醚,继续搅拌直至反应结束,将反应结束后的液体,减压蒸馏除去溶剂,得到透明油状的中间产物1:
[0094][0095]
步骤二、将步骤一中收得的中间产物1用20ml苯溶解,加入3g中性氧化铝,于室温条件下搅拌,直至反应结束,抽滤除去中性氧化铝,用二氯甲烷冲洗滤膜上的中性氧化铝得到滤液,将滤液减压抽滤,得到油状物,将油状物采用乙酸乙酯与石油醚体积比为1:5的硅胶柱层析分离得到443mg透明油状物,即中间产物2:
[0096][0097]
产率为53%,经核磁共振和质谱分析确定中间产物2的结构;
[0098]
步骤三、将175mg(0.42mmol)中间产物2用30ml无水乙醇搅拌溶解,加热至80℃条件下,在加热过程中加入115mg(0.84mmol)三水乙酸钠并搅拌,加入59mg(0.84mmol)盐酸羟胺,然后回流搅拌直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅰ,粗产物ⅰ采用乙酸乙酯与石油醚体积比为1:3的硅胶柱层析分离得到154mg白色固体,即中间产物3:
[0099][0100]
产率为85%,通过熔点测定实验得到熔点为:145~147℃,经核磁共振和质谱分析确定中间产物3的结构;
[0101]
步骤四、将325mg(0.75mmol)中间产物3用20ml无水乙醇加热搅拌溶解后,依次加入187mg(3.00mmol)氰基硼氢化钠、203mg(0.75mmol)五氯化钼和308mg(2.25mmol)一水合硫酸氢钠,于室温条件下继续搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅱ,粗产物ⅱ采用乙酸乙酯与石油醚体积比为1:15的硅胶柱层析分离得到252mg淡黄色固体,即中间产物4:
[0102][0103]
产率为80%,通过熔点测定实验得到熔点为:121~123℃,经核磁共振和质谱分析确定中间产物4的结构;
[0104]
步骤五、将250mg(0.596mmol)中间产物4用15ml二氯甲烷搅拌溶解后,加入250μl(1.788mmol)三乙胺后,于0℃条件下搅拌15min,滴加98μl(0.76mmol)5-氯戊酰氯,继续搅拌30min后于室温条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅲ,粗产物ⅲ采用乙酸乙酯与石油醚体积比为1:4的硅胶柱层析分离得到233mg白色固体,即中间产物5c:
[0105][0106]
产率为73%,通过熔点测定实验得到熔点为:152~153℃,经核磁共振和质谱分析确定中间产物5c的结构;
[0107]
步骤六、将150mg(0.279mmol)中间产物5c用15ml n,n-二甲基甲酰胺搅拌溶解后,加入1.5mol/l 1ml的硒氰酸钾溶液,通入氩气,并用锡纸包裹,于80℃条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅳ,粗产物ⅳ采用乙酸乙酯与石油醚体积比为1:2的硅胶柱层析分离得到40mg白色固体,即b-降-3,5-二羟基胆甾-6-硒氰基戊酰胺(6c):
[0108][0109]
产率为23%,通过熔点测定实验得到熔点为:167~168℃,经核磁共振和质谱分析确定b-降-3,5-二羟基胆甾-6-硒氰基戊酰胺的结构。
[0110]
《实施例4》
[0111]
b-降-3,5-二羟基胆甾-6-对硒氰基甲基苯甲酰胺(6d)的制备:
[0112]
步骤一、将772mg(2mmol)胆固醇用20ml二氯甲烷和5ml甲醇溶解,在-78℃以下通入臭氧,并搅拌反应,经薄层层析技术监测胆固醇反应完全后,停止通入臭氧,加入2ml二甲硫醚,继续搅拌直至反应结束,将反应结束后的液体,减压蒸馏除去溶剂,得到透明油状的中间产物1:
[0113][0114]
步骤二、将步骤一中收得的中间产物1用20ml苯溶解,加入3g中性氧化铝,于室温条件下搅拌,直至反应结束,抽滤除去中性氧化铝,用二氯甲烷冲洗滤膜上的中性氧化铝得到滤液,将滤液减压抽滤,得到油状物,将油状物采用乙酸乙酯与石油醚体积比为1:5的硅胶柱层析分离得到443mg透明油状物,即中间产物2:
[0115][0116]
产率为53%,经核磁共振和质谱分析确定中间产物2的结构;
[0117]
步骤三、将175mg(0.42mmol)中间产物2用30ml无水乙醇搅拌溶解,加热至80℃条件下,在加热过程中加入115mg(0.84mmol)三水乙酸钠并搅拌,加入59mg(0.84mmol)盐酸羟胺,然后回流搅拌直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅰ,粗产物ⅰ采用乙酸乙酯与石油醚体积比为1:3的硅胶柱层析分离得到154mg白色固体,即中间产物3:
[0118][0119]
产率为85%,通过熔点测定实验得到熔点为:145~147℃,经核磁共振和质谱分析确定中间产物3的结构;
[0120]
步骤四、将325mg(0.75mmol)中间产物3用20ml无水乙醇加热搅拌溶解后,依次加入187mg(3.00mmol)氰基硼氢化钠、203mg(0.75mmol)五氯化钼和308mg(2.25mmol)一水合硫酸氢钠,于室温条件下继续搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅱ,粗产物ⅱ采用乙酸乙酯与石油醚体积比为1:15的硅胶柱层析分离得到252mg淡黄色固体,即中间产物4:
[0121][0122]
产率为80%,通过熔点测定实验得到熔点为:121~123℃,经核磁共振和质谱分析确定中间产物4的结构;
[0123]
步骤五、将250mg(0.596mmol)中间产物4用15ml二氯甲烷搅拌溶解后,加入250μl(1.788mmol)三乙胺后,于0℃条件下搅拌15min,滴加111μl(0.76mmol)4-氯甲基苯甲酰氯,继续搅拌30min后于室温条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅲ,粗产物ⅲ采用乙酸乙酯与石油醚体积比为1:4的硅胶柱层析分离得到170mg白色固体,即中间产物5d:
[0124][0125]
产率为50%,通过熔点测定实验得到熔点为:190~191℃,经核磁共振和质谱分析确定中间产物5d的结构;
[0126]
步骤六、将170mg(0.298mmol)中间产物5d用15ml n,n-二甲基甲酰胺搅拌溶解后,加入1.5mol/l 1ml的硒氰酸钾溶液,通入氩气,并用锡纸包裹,于80℃条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅳ,粗产物ⅳ采用乙酸乙酯与石油醚体积比为1:2的硅胶柱层析分离得到158mg白色固体,即b-降-3,5-二羟基胆甾-6-对硒氰基甲基苯甲酰胺(6d):
[0127][0128]
产率为82%,通过熔点测定实验得到熔点为:174~176℃,经核磁共振和质谱分析确定b-降-3,5-二羟基胆甾-6-对硒氰基甲基苯甲酰胺的结构。
[0129]
《实施例5》
[0130]
b-降-3,5-二羟基胆甾-6-间硒氰基甲基苯甲酰胺(6e)的制备:
[0131]
步骤一、将772mg(2mmol)胆固醇用20ml二氯甲烷和5ml甲醇溶解,在-78℃以下通入臭氧,并搅拌反应,经薄层层析技术监测胆固醇反应完全后,停止通入臭氧,加入2ml二甲硫醚,继续搅拌直至反应结束,将反应结束后的液体,减压蒸馏除去溶剂,得到透明油状的中间产物1:
[0132][0133]
步骤二、将步骤一中收得的中间产物1用20ml苯溶解,加入3g中性氧化铝,于室温条件下搅拌,直至反应结束,抽滤除去中性氧化铝,用二氯甲烷冲洗滤膜上的中性氧化铝得到滤液,将滤液减压抽滤,得到油状物,将油状物采用乙酸乙酯与石油醚体积比为1:5的硅胶柱层析分离得到443mg透明油状物,即中间产物2:
[0134][0135]
产率为53%,经核磁共振和质谱分析确定中间产物2的结构;
[0136]
步骤三、将175mg(0.42mmol)中间产物2用30ml无水乙醇搅拌溶解,加热至80℃条件下,在加热过程中加入115mg(0.84mmol)三水乙酸钠并搅拌,加入59mg(0.84mmol)盐酸羟胺,然后回流搅拌直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅰ,粗产物ⅰ采用乙酸乙酯与石油醚体积比为1:3的硅胶柱层析分离得到154mg白色固体,即中间产物3:
[0137][0138]
产率为85%,通过熔点测定实验得到熔点为:145~147℃,经核磁共振和质谱分析确定中间产物3的结构;
[0139]
步骤四、将325mg(0.75mmol)中间产物3用20ml无水乙醇加热搅拌溶解后,依次加入187mg(3.00mmol)氰基硼氢化钠、203mg(0.75mmol)五氯化钼和308mg(2.25mmol)一水合硫酸氢钠,于室温条件下继续搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅱ,粗产物ⅱ采用乙酸乙酯与石油醚体积比为1:15的硅胶柱层析分离得到252mg淡黄色固体,即中间产物4:
[0140][0141]
产率为80%,通过熔点测定实验得到熔点为:121-123℃,经核磁共振和质谱分析确定中间产物4的结构;
[0142]
步骤五、将250mg(0.596mmol)中间产物4用15ml二氯甲烷搅拌溶解后,加入250μl(1.788mmol)三乙胺后,于0℃条件下搅拌15min,滴加111μl(0.76mmol)3-氯甲基苯甲酰氯,继续搅拌30min后于室温条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅲ,粗产物ⅲ采用乙酸乙酯与石油醚体积比为1:4的硅胶柱层析分离得到259mg白色固体,即中间产物5e:
[0143][0144]
产率为76%,通过熔点测定实验得到熔点为:173~174℃,经核磁共振和质谱分析确定中间产物5e的结构;
[0145]
步骤六、将168mg(0.294mmol)中间产物5e用15ml n,n-二甲基甲酰胺搅拌溶解后,加入1.5mol/l 1ml的硒氰酸钾溶液,通入氩气,并用锡纸包裹,于80℃条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅳ,粗产物ⅳ采用乙酸乙酯与石油醚体积比为1:2的硅胶柱层析分离得到98mg白色固体,即b-降-3,5-二羟基胆甾-6-间硒氰基甲基苯甲酰胺(6e):
[0146][0147]
产率为50%,通过熔点测定实验得到熔点为:168~170℃,经核磁共振和质谱分析确定b-降-3,5-二羟基胆甾-6-间硒氰基甲基苯甲酰胺的结构。
[0148]
《实施例6》
[0149]
b-降-3,5-二羟基胆甾-6-硒氰基庚酰胺(6f)的制备:
[0150]
步骤一、将772mg(2mmol)胆固醇用20ml二氯甲烷和5ml甲醇溶解,在-78℃以下通入臭氧,并搅拌反应,经薄层层析技术监测胆固醇反应完全后,停止通入臭氧,加入2ml二甲硫醚,继续搅拌直至反应结束,将反应结束后的液体,减压蒸馏除去溶剂,得到透明油状的中间产物1:
[0151][0152]
步骤二、将步骤一中收得的中间产物1用20ml苯溶解,加入3g中性氧化铝,于室温条件下搅拌,直至反应结束,抽滤除去中性氧化铝,用二氯甲烷冲洗滤膜上的中性氧化铝得到滤液,将滤液减压抽滤,得到油状物,将油状物采用乙酸乙酯与石油醚体积比为1:5的硅胶柱层析分离得到443mg透明油状物,即中间产物2:
[0153][0154]
产率为53%,经核磁共振和质谱分析确定中间产物2的结构;
[0155]
步骤三、将175mg(0.42mmol)中间产物2用30ml无水乙醇搅拌溶解,加热至80℃条件下,在加热过程中加入115mg(0.84mmol)三水乙酸钠并搅拌,加入59mg(0.84mmol)盐酸羟胺,然后回流搅拌直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅰ,粗产物ⅰ采用乙酸乙酯与石油醚体积比为1:3的硅胶柱层析分离得到154mg白色固体,即中间产物3:
[0156][0157]
产率为85%,通过熔点测定实验得到熔点为:145~147℃,经核磁共振和质谱分析确定中间产物3的结构;
[0158]
步骤四、将325mg(0.75mmol)中间产物3用20ml无水乙醇加热搅拌溶解后,依次加入187mg(3.00mmol)氰基硼氢化钠、203mg(0.75mmol)五氯化钼和308mg(2.25mmol)一水合硫酸氢钠,于室温条件下继续搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅱ,粗产物ⅱ采用乙酸乙酯与石油醚体积比为1:15的硅胶柱层析分离得到252mg淡黄色固体,即中间产物4:
[0159][0160]
产率为80%,通过熔点测定实验得到熔点为:121~123℃,经核磁共振和质谱分析确定中间产物4的结构;
[0161]
步骤五、在氩气保护条件下,往150mg(0.358mmol)中间产物4中依次加入75mg(0.358mmol)的7-溴庚酸、187mg(0.358mmol)六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷、22mg(0.179mmol)4-二甲氨基吡啶、150μl(1.074mmol)三乙胺以及10ml二氯甲烷,于室温条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅲ,粗产物ⅲ采用乙酸乙酯与石油醚体积比为1:3的硅胶柱层析分离得到119mg白色固体,即中间产物5f:
[0162][0163]
产率为54%,通过熔点测定实验得到熔点为:112~114℃,经核磁共振和质谱分析确定中间产物5f的结构;
[0164]
步骤六、将80mg(0.131mmol)中间产物5f用10ml n,n-二甲基甲酰胺搅拌溶解后,加入1.5mol/l 1ml的硒氰酸钾溶液,通入氩气,并用锡纸包裹,于80℃条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅳ,粗产物ⅳ采用乙酸乙酯与石油醚体积比为1:2的硅胶柱层析分离得到53mg白色固体,即b-降-3,5-二羟基胆甾-6-硒氰基庚酰胺(6f):
[0165][0166]
产率为64%,通过熔点测定实验得到熔点为:114~115℃,经核磁共振和质谱分析确定b-降-3,5-二羟基胆甾-6-硒氰基庚酰胺的结构。
[0167]
《实施例7》
[0168]
b-降-3,5-二羟基胆甾-6-硒氰基丁酰胺硒氰基十一酰胺(6g)的制备:
[0169]
步骤一、将772mg(2mmol)胆固醇用20ml二氯甲烷和5ml甲醇溶解,在-78℃以下通入臭氧,并搅拌反应,经薄层层析技术监测胆固醇反应完全后,停止通入臭氧,加入2ml二甲硫醚,继续搅拌直至反应结束,将反应结束后的液体,减压蒸馏除去溶剂,得到透明油状的中间产物1:
[0170][0171]
步骤二、将步骤一中收得的中间产物1用20ml苯溶解,加入3g中性氧化铝,于室温条件下搅拌,直至反应结束,抽滤除去中性氧化铝,用二氯甲烷冲洗滤膜上的中性氧化铝得到滤液,将滤液减压抽滤,得到油状物,将油状物采用乙酸乙酯与石油醚体积比为1:5的硅胶柱层析分离得到443mg透明油状物,即中间产物2:
[0172][0173]
产率为53%,经核磁共振和质谱分析确定中间产物2的结构;
[0174]
步骤三、将175mg(0.42mmol)中间产物2用30ml无水乙醇搅拌溶解,加热至80℃条件下,在加热过程中加入115mg(0.84mmol)三水乙酸钠并搅拌,加入59mg(0.84mmol)盐酸羟胺,然后回流搅拌直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅰ,粗产物ⅰ采用乙酸乙酯与石油醚体积比为1:3的硅胶柱层析分离得到154mg白色固体,即中间产物3:
[0175][0176]
产率为85%,通过熔点测定实验得到熔点为:145~147℃,经核磁共振和质谱分析确定中间产物3的结构;
[0177]
步骤四、将325mg(0.75mmol)中间产物3用20ml无水乙醇加热搅拌溶解后,依次加入187mg(3.00mmol)氰基硼氢化钠、203mg(0.75mmol)五氯化钼和308mg(2.25mmol)一水合硫酸氢钠,于室温条件下继续搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅱ,粗产物ⅱ采用乙酸乙酯与石油醚体积比为1:15的硅胶柱层析分离得到252mg淡黄色固体,即中间产物4:
[0178][0179]
产率为80%,通过熔点测定实验得到熔点为:121~123℃,经核磁共振和质谱分析确定中间产物4的结构;
[0180]
步骤五、在氩气保护条件下,往150mg(0.358mmol)中间产物4中依次加入95mg(0.358mmol)的溴代十一酸、187mg(0.358mmol)六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷、22mg(0.179mmol)4-二甲氨基吡啶、150μl(1.074mmol)三乙胺以及10ml二氯甲烷,于室温条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅲ,粗产物ⅲ采用乙酸乙酯与石油醚体积比为1:3的硅胶柱层析分离得到159mg白色固体,即中间产物5g:
[0181][0182]
产率为67%,通过熔点测定实验得到熔点为:87~88℃,经核磁共振和质谱分析确定中间产物5g的结构;
[0183]
步骤六、将200mg(0.300mmol)中间产物5g用10ml n,n-二甲基甲酰胺搅拌溶解后,加入1.5mol/l 1ml的硒氰酸钾溶液,通入氩气,并用锡纸包裹,于80℃条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅳ,粗产物ⅳ采用乙酸乙酯与石油醚体积比为1:2的硅胶柱层析分离得到166mg白色固体,即b-降-3,5-二羟基胆甾-6-硒氰基十一酰胺(6g):
[0184][0185]
产率为80%,通过熔点测定实验得到熔点为:91~92℃,经核磁共振和质谱分析确定b-降-3,5-二羟基胆甾-6-硒氰基十一酰胺的结构。
[0186]
《实施例8》
[0187]
b-降-3,5-二羟基胆甾-6-硒氰乙氧基苯甲酰胺(6h)的制备:
[0188]
步骤一、在304mg(2mmol)对羟基苯甲酸甲酯中加入830mg(6mmol)碳酸钾和15ml乙腈,于回流条件下并搅拌反应15min,然后加入527μl(6mmol)1,2-二溴乙烷,继续搅拌直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物,粗产物采用乙酸乙酯与石油醚体积比为1:20的硅胶柱层析分离得到662mg白色固体,即中间产物7:
[0189][0190]
产率为64%,通过熔点测定实验得到熔点为:70~71℃,经核磁共振和质谱分析确定中间产物7的结构;
[0191]
步骤二、将306mg(1.186mmol)中间产物7溶于10ml四氢呋喃中,加入lioh水溶液
(其中57mg lioh、2.4ml水),于65℃下搅拌直至反应结束,将反应后的液体用10wt%hcl调至ph=2~3后,经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物,粗产物采用乙酸乙酯与石油醚体积比为1:7的硅胶柱层析分离得到得到150mg白色固体,即4-(2-溴乙氧基)苯甲酸(8):
[0192][0193]
产率为52%,通过熔点测定实验得到熔点为:181~183℃,经核磁共振和质谱分析确定4-(2-溴乙氧基)苯甲酸的结构;
[0194]
步骤三、在氩气保护条件下,往200mg(0.477mmol)中间产物4中依次加入116mg(0.477mmol)的4-(2-溴乙氧基)苯甲酸、248mg(0.477mmol)六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷、29mg(0.239mmol)4-二甲氨基吡啶、199μl(1.431mmol)三乙胺以及10ml二氯甲烷,于室温条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物
ⅴ
,粗产物
ⅴ
采用乙酸乙酯与石油醚体积比为1:4的硅胶柱层析分离得到150mg白色固体,即中间产物5h:
[0195][0196]
产率为49%,通过熔点测定实验得到熔点为:143~144℃,经核磁共振和质谱分析确定中间产物5h的结构;
[0197]
步骤四、将120mg(0.186mmol)中间产物5h用10ml n,n-二甲基甲酰胺搅拌溶解后,加入1.5mol/l 1ml的硒氰酸钾溶液,通入氩气,并用锡纸包裹,于80℃条件下搅拌,直至反应结束,将反应结束后的液体经乙酸乙酯萃取得到有机相,有机相经饱和氯化钠、饱和碳酸氢钠溶液洗涤、无水硫酸钠干燥、浓缩后得到粗产物ⅵ,粗产物ⅵ采用乙酸乙酯与石油醚体积比为1:2的硅胶柱层析分离得到60mg白色固体,即b-降-3,5-二羟基胆甾-6-硒氰乙氧基苯甲酰胺(6h):
[0198][0199]
产率为48%,通过熔点测定实验得到熔点为:196~197℃,经核磁共振和质谱分析确定b-降-3,5-二羟基胆甾-6-硒氰乙氧基苯甲酰胺的结构。
[0200]
《体外抗肿瘤活性实验》
[0201]
采用mts法研究雌二醇硒氰酸酯类化合物6a~6h以及它们对应的前体中间产物5a~5h对人宫颈癌细胞(hela)、人乳腺癌细胞(sk-ov-3)、人肝癌细胞(hepg-2)、人乳腺癌细胞(t47d和mcf-7)的抗肿瘤活性。
[0202]
在培养有对数生长期细胞的96孔酶标板中加入相同浓度的胆固醇硒氰酸酯类化合物作为实验组,并选择市售药物阿比特龙作为阳性对照组,在相同条件下设置空白组,每种处理均设置4个复孔。培养72h后加入20μl mtt(5mg/ml),继续孵育4h。吸取上清液,加入200μl的二甲基亚砜,将其置于摇床上约10min充分混匀,最后用酶标仪在波长为490nm处测定其吸光度od,根据以下公式计算各组细胞的抑制率:
[0203][0204]
根据各组细胞的抑制率使用graphpad prism 8软件计算其半数存活浓度ic
50
值并分析统计结果如表1所述所示:
[0205]
表1胆固醇硒氰酸酯类化合物的体外抗肿瘤活性的ic
50
值(单位:μmol/l)
[0206][0207][0208]
由表1数据可知,本发明公开的大部分胆固醇硒氰酸酯类化合物抗肿瘤细胞增殖活性均优于它们的前体;从构效关系上来看,当连接的直链烃基链长从2个碳增长到5时,化合物6c表现出最好的抑制活性,但随着链长的增长,抑制效果随之减弱;连接直链烃基与苯
环苄基的抑制作用差别不大,但它们的作用效果均明显高于市售抗肿瘤药物阿比特龙。本发明公开的胆固醇硒氰酸酯化合物对几种肿瘤细胞具有很好的抑制作用,如化合物6c、6d和6h对人卵巢癌细胞(sk-ov-3)的ic
50
值分别为4.79、3.39和4.16μmol/l,化合物6c、6d和6h对乳腺癌细胞(t47d)细胞的ic
50
值分别为8.73、6.67和5.34μmol/l,化合物6c、6h对所有受试肿瘤细胞均具有明显的抑制作用,ic
50
值均小于9μmol/l,且比市售药物阿比特龙对肿瘤细胞的抑制作用更加优异。
[0209]
尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用,它完全可以被适用于各种适合本发明的领域,对于熟悉本领域的人员而言,可容易地实现另外的修改,因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节和这里示出与描述的实施例。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。