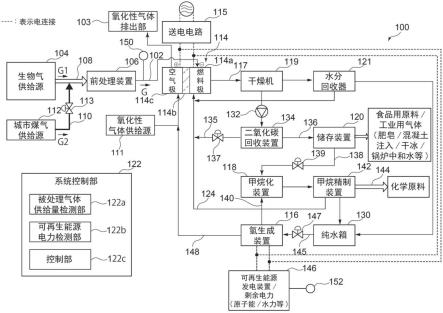
1.本发明属于医药技术领域,具体涉及一种氮唑类抗真菌化合物及其应用。
背景技术:
2.近年来,随着器官移植的普遍开展和hiv感染人群的不断扩大,造成了低免疫力人群的增多,加以侵入性诊疗设备的使用,导致了侵袭性深部真菌感染在全球蔓延。与日趋严重的真菌感染相比,抗真菌新药的研发速度远不及真菌产生耐药的速度,而日益严重的真菌耐药性也是导致免疫缺陷病人侵袭性真菌感染的发病率和死亡率逐年升高的重要原因之一。目前可用于临床的品种主要包括多烯类、氮唑类以及棘白菌素类。而三氮唑类药物是一类最具有发展潜力和优势的药物,先后有伊曲康唑(1989)、氟康唑(1990)、伏立康唑(2001)、泊沙康唑(2006)、艾沙康唑(2015)等药物上市,已成为临床抗真菌感染的一线药物。但是,由于唑类抗真菌药物的长期治疗和重复给药过程真菌对其产生了耐药性,使临床治疗失败。因此,开发新一代抗真菌尤其是抗耐药真菌的药物仍十分重要。
技术实现要素:
3.本发明的目的是要提供一种氮唑类抗真菌化合物及其应用。
4.本发明解决上述技术问题的技术方案如下:
5.一种氮唑类抗真菌化合物、其光学异构体或其药学上可接受的盐,其特征在于,所述化合物包括通式(i)或(ii),所述通式(i)或(ii)化合物含有三唑或四唑及二氟苯基的结构,
[0006][0007]
其中,x选自c原子或n原子;
[0008]
y选自甲基或h原子;
[0009]
z选自-nh-或o原子;
[0010]
r选自3,4-(亚甲二氧基)苯乙基、3,4-(亚甲二氧基)苯甲基、3,4-(亚甲二氧基)苯基、3-[3,4-(亚甲二氧基)苯基]丙烯酰基、3-[3,4-(亚甲二氧基)苯基]丙酰基、3-(2,2-二氟苯并[d][1,3]二氧杂环戊烷-5-基)丙烯酰基、1,4-苯并二噁烷-6-丙烯酰基、3,4-(亚甲二氧基)苯乙酰基、3-胡椒酰基、2-胡椒酰基、2,3-二氢苯并呋喃-5-乙酰基、1-(2,2-二氟苯并[d][1,3]二氧杂环戊烯-5-基)环丙烷甲酰基、(2,3-二氢-苯并[1,4]二氧-6)-乙酰基、2,3-二氢-1,4-苯并二噁烷-6-酰基、2,3-二氢-1,4-苯并二噁烷-5-酰基、3-(2,3-二氢苯并呋喃-5-基)丙烯酰基、3,4-二羟基肉桂酰基、3-羟基-4-甲氧基肉桂酰基、4-羟基-3-甲氧基肉
桂酰基、3,4-二甲氧基肉桂酰基;*标识的碳原子为手性碳原子或非手性碳原子。
[0011]
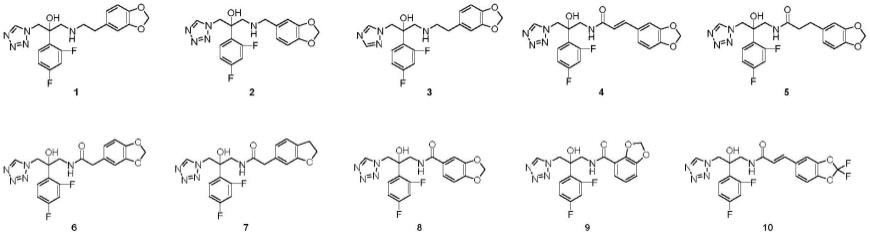
本发明具有代表性的优选化合物结构,1hnmr及ms表征数据见表1。
[0012]
表1优选化合物的结构及表征数据
[0013]
[0014]
[0015]
[0016]
[0017]
[0018]
[0019][0020]
优选的所述的一种氮唑类抗真菌化合物、其光学异构体或其药学上可接受的盐,所述化合物(i)或(ii)包括异构体中的任何一种或混合物。
[0021]
优选的所述的一种氮唑类抗真菌化合物、其光学异构体或其药学上可接受的盐,所述的药学上可接受的盐是盐酸盐、醋酸盐、草酸盐、硫酸盐、硫酸氢盐、氢溴酸盐、甲磺酸盐或柠檬酸盐。
[0022]
本发明还提供一种氮唑类抗真菌化合物、其光学异构体或其药学上可接受的盐在制备抗真菌药物中的应用。
[0023]
本发明中化合物的中文命名与结构式有冲突的,以结构式为准;结构式有明显错误的除外。
[0024]
本发明的有益效果在于:本发明公开了一种氮唑类抗真菌化合物,具体涉及具有化学结构通式(i)和(ii)的氮唑类抗真菌化合物和光学异构体及其药学上可接受的盐,可以用于制备新的抗真菌药物。本发明的化合物与临床常用药氟康唑相比对致病真菌白色念珠菌具有很强的抑菌活性。药效团融合是发现新型药物的重要策略,本发明将小檗碱衍生物的关键药效团胡椒基及其类似物与氮唑类抗真菌药物的药效团融合,设计合成了系列可用于抗真菌的化合物。本发明的化合物大部分具有良好的抗真菌活性特点,部分化合物对白色念珠菌的抑制优于阳性药氟康唑,具有优秀的抗真菌活性。
具体实施方式
[0025]
以下结合实例说明本发明,但不限制本发明。在本领域内,技术人员对本发明所做的简单替换或改进均属于本发明所保护的技术方案内。
[0026]
以下提供的方法实施例用于促进对本发明的制备方法的进一步了解,使用的具体物质、种类和条件确定为是对本发明的进一步说明,并不是对其合理范围的限制。在下面描述的使用的原料、试剂或者可以市场购买到,或者可以由本领域普通技术人员轻易制备得到。
[0027]
本发明通式(i)的部分化合物可以按照合成路线(1)的方法制备。
[0028]
合成路线(1):
[0029][0030]
实施例1:1-((2-(苯并[d][1,3]二氧戊环-5-基)乙基)氨基)-2-(2,4-二氟苯基)-3-(1h-四唑-1-基)丙-2-醇的制备[(i)a化合物1]
[0031]
第一步:2-(1h-1,2,3,4-四唑-1-基)-2',4'-二氟苯乙酮的制备类[(2)类中间体]
[0032]
将1h-四氮唑2.74g(0.065mol)溶于100ml的n,n-二甲基甲酰胺(dmf)圆底烧瓶中,置于0℃的冰水浴下搅拌混匀,后分批加入60%nah 1.56g(0.065mol),在冰浴下搅拌0.5h后,加入原料(1)2'-氯-2,4-二氟苯乙酮5.7g(0.05mol),反应在室温下搅拌2h,将反应液倒入冰水中,析出大量固体,过滤得黄色固体,滤液用乙酸乙酯(100ml*2)萃取两遍,合并有机层,用饱和食盐水(150ml*3)洗涤三遍,无水硫酸钠干燥,过滤,减压浓缩得固体,合并两次所得固体,硅胶柱纯化(洗脱剂:ea:pe=1:1.5),得7.2g类白色固体,收率64.2%。该化合物结构经核磁共振氢谱(1h nmr)和lc-ms鉴定,其数据如下:1h nmr(400mhz,dmso-d6)δ9.33(s,1h),8.08(td,j=8.7,6.6hz,1h),7.58(ddd,j=11.7,9.3,2.5hz,1h),7.34(td,j=8.5,2.5hz,1h),6.18(d,j=3.0hz,2h).ms(esi)m/z:225.20[m h]
,223.10[m-h]-。
[0033]
第二步:1-[2-(2,4-二氟苯基)-2,3-环氧丙基]-1h-1,2,3,4-四唑的制备[(3)类中间体]
[0034]
向2-(1h-1,2,3,4-四唑-1-基)-2',4'-二氟苯乙酮[(2)类中间体](1.12g,5mmol)的10ml二氯甲烷溶液中,加入三甲基碘化亚砜2.01g(5mmol)和10ml的20%氢氧化钠(2g,50mmol)溶液中。将混合物微波反应5h,微波反应器设定温度40℃,最大输出功率为50w。反应结束后,用乙酸乙酯(20ml*3)萃取混合物,并用饱和食盐水(20ml*3)洗涤有机层三遍,无水硫酸钠干燥,减压浓缩得黄色油状物,硅胶柱纯化(洗脱剂:ea:pe=1:1.5),得0.9g淡红色油状物(收率为75%)。该化合物结构经核磁共振氢谱(1h nmr)和lc-ms鉴定,其数据如下:1h nmr(400mhz,dmso-d6)δ9.35(s,1h),7.32(d,j=11.0hz,1h),7.22(td,j=8.5,6.7hz,1h),7.04(td,j=8.5,2.0hz,1h),5.12(d,j=14.9hz,1h),4.89(d,j=14.9hz,1h),3.20(d,j=4.6hz,1h),3.04(d,j=4.6hz,1h).ms(esi),m/z:239.20[m h]
,237.10[m-h]-。
[0035]
第三步:1-((2-(苯并[d][1,3]二氧戊环-5-基)乙基)氨基)-2-(2,4-二氟苯基)-3-(1h-四唑-1-基)丙-2-醇的制备[(i)a化合物1]
[0036]
在50ml的圆底烧瓶中加入1-[2-(2,4-二氟苯基)-2,3-环氧丙基]-1h-1,2,3,4-四唑[(3)类中间体](70mg,2.95mmol),k2co3(53mg,3.8mmol)和10ml的n-甲基吡咯烷酮(nmp),搅拌混匀,并滴加3,4-亚甲二氧基苯乙胺溶液(49mg,3.54mmol),80℃反应5h,tlc检测反应的进行,反应完成后,冷却至室温,乙酸乙酯(10ml*3)萃取混合物三遍,并用饱和食盐水
(10ml*3)洗涤有机层三遍,无水硫酸钠干燥,减压浓缩得粗品,硅胶柱纯化(洗脱剂:dcm:meoh=15:1),得71mg白色固体(收率为60%)。该化合物核磁共振氢谱(1h nmr)和lc-ms表征数据见表1。
[0037]
(i)a化合物2,参照具体实施例1的第三步将3,4-亚甲二氧基苯甲胺代替3,4-亚甲二氧基苯乙胺制备。该化合物核磁共振氢谱(1hnmr)和lc-ms表征数据见表1。
[0038]
(i)a化合物3,参照具体实施例1将第一步将1h-1,2,4-三氮唑代替1h-四氮唑,后续步骤替换为相应的中间体参照具体实施例1制备。该化合物核磁共振氢谱(1hnmr)和lc-ms表征数据见表1。
[0039]
实施例2:(e)-3-(苯并[d][1,3]二氧戊环-5-基)-n-(2-(2,4-二氟苯基)-2-羟基-3-(1h-四唑-1-基))丙基)丙烯酰胺的制备[(i)b化合物4]
[0040]
第一步:1-氨基-2-(2,4-二氟苯基)-3-(1h-四唑-1-基)丙-2-醇的制备[(4)类中间体]
[0041]
将1-[2-(2,4-二氟苯基)-2,3-环氧丙基]-1h-1,2,3,4-四唑(0.9g,3.8mmol)[(3)类中间体]和8ml的7m氨的甲醇溶液加入到耐压管中,85℃反应2h,待冷却后减压浓缩,硅胶柱纯化(洗脱剂dcm:meoh=10:1),得0.87g固体(收率为90%)。该化合物结构经核磁共振氢谱(1h nmr)和lc-ms鉴定,其数据如下:1h nmr(400mhz,dmso-d6)δ9.13(s,1h),7.28(td,j=9.0,6.8hz,1h),7.25
–
7.18(m,1h),6.95(td,j=8.5,2.6hz,1h),4.84(q,j=14.3hz,2h),3.04
–
2.94(m,2h),1.91(s,3h).ms(esi),m/z:256.31[m h]
,254.25[m-h]-。
[0042]
第二步:(e)-3-(苯并[d][1,3]二氧戊环-5-基)-n-(2-(2,4-二氟苯基)-2-羟基-3-(1h-四唑-1-基))丙基)丙烯酰胺的制备[(i)b化合物4]
[0043]
向25ml的圆底烧瓶中加入1-氨基-2-(2,4-二氟苯基)-3-(1h-四唑-1-基)丙-2-醇[(4)类中间体](80mg,3.1mmol),3,4-(亚甲二氧)肉桂酸(66mg,3.4mmol)和5ml的dmf搅拌,加入hatu(155mg,4.0mmol),滴加n,n二异丙基乙胺(diea,150μl,9.3mmol),室温搅拌反应4h,反应完成后用乙酸乙酯(10ml*3)萃取混合物三遍,并用饱和食盐水(10ml*3)洗涤有机层三遍,无水硫酸钠干燥,减压浓缩得粗品,硅胶柱纯化(洗脱剂:ea:pe=2:1),得87mg白色固体(收率为65%)。该化合物核磁共振氢谱(1h nmr)和lc-ms表征数据见表1。
[0044]
(i)b化合物5~21可选用相应的羧酸,参照具体实施例2第二步通过酰胺缩合的方法制备。
[0045]
本发明通式(i)的部分化合物可以按照合成路线(2)的方法制备。
[0046]
合成路线(2):
[0047][0048]
实施例3:(e)-3-(苯并[d][1,3]二氧戊环-5-基)-n-((2r,3r)-3-(2,4-二氟苯基)-3-羟基-4-(1h-1,2,4-三唑-1-基))丁-2-基)丙烯酰胺的制备(化合物22)
[0049]
第一步:(2r,3r)-3-氨基-2-(2,4-二氟苯基)-1-(1h-1,2,4-三唑-1-基)丁-2-醇[中间体(6)]
[0050]
称取原料(5)1-(((2r,3s)-2-(2,4-二氟苯基)-3-甲基环氧乙烷-2-基)甲基)-1h-1,2,4-三唑(2.0g,8mmol),叠氮化钠(776mg,12mmol)和氯化铵(553mg,10.4mmol)加入到100ml圆底烧瓶中,并加入20ml的dmf,80℃搅拌反应10h,待反应完成后,加入20ml的水,用乙酸乙酯(20ml*3)萃取,合并有机层,用饱和食盐水(20ml*3)洗涤三遍,并用无水硫酸钠干燥,减压浓缩得油状物。得到得油状物加入到50ml的圆底烧瓶中,加入10ml甲醇溶解,并加入200mg的10%的pd/c,氢气置换气体三遍,在室温下反应5h后,减压浓缩得粗品,硅胶柱纯化(洗脱剂dcm:meoh=10:1),得到1.41g白色固体(产率为66%)。该化合物结构经核磁共振氢谱(1h nmr)和lc-ms鉴定,其数据如下:1h nmr(400mhz,dmso-d6)δ8.26(s,1h),7.64(s,1h),7.26(td,j=8.9,7.2hz,1h),7.12(ddd,j=11.9,9.2,2.5hz,1h),6.87(td,j=8.5,2.5hz,1h),4.66(q,j=14.4hz,2h),1.90(s,2h),0.70(d,j=6.5hz,3h).m/z:269.4[m h]
,267.1[m-h]-。
[0051]
第二步:(e)-3-(苯并[d][1,3]二氧戊环-5-基)-n-((2r,3r)-3-(2,4-二氟苯基)-3-羟基-4-(1h-1,2,4-三唑-1-基))丁-2-基)丙烯酰胺的制备(化合物22)
[0052]
向25ml的圆底烧瓶中加入(2r,3r)-3-氨基-2-(2,4-二氟苯基)-1-(1h-1,2,4-三唑-1-基)丁-2-醇[中间体(6)](83mg,3.1mmol),3,4-(亚甲二氧)肉桂酸(66mg,3.4mmol)和5ml的dmf搅拌,并其中加入hatu(155mg,4.0mmol),滴加n,n二异丙基乙胺(diea,150μl,9.3mmol),室温搅拌反应4h,反应完成后用乙酸乙酯(10ml*3)萃取混合物三遍,并用饱和食盐水(10ml*3)洗涤有机层三遍,无水硫酸钠干燥,减压浓缩得粗品,硅胶柱纯化(洗脱剂:ea:pe=2:1),得87mg白色固体(收率为65%)。该化合物核磁共振氢谱(1hnmr)和lc-ms表征数据见表1。
[0053]
(i)c化合物23~30可选用相应的羧酸,参照具体实施例3第二步通过酰胺缩合的方法制备。
[0054]
本发明通式(ii)的化合物可以按照合成路线(3)的方法制备。
[0055]
合成路线(3):
[0056][0057]
实施例4:1-((3r,5r)-(5-((1h-1,2,4-三唑-1-基)甲基)-5-(2,4-二氟苯基)四氢呋喃-3-基)-n-(苯并[d][1,3]二氧戊环-5-基甲基)甲胺的制备(化合物31)
[0058]
第一步:((3s,5r)-5-((1h-1,2,4-三唑-1-基)甲基)-5-(2,4-二氟苯基)四氢呋喃-3-基)4-甲基苯磺酸甲酯的制备[中间体(8)]
[0059]
将5.9g原料(7)((3r,5r)-5-((1h-1,2,4-三唑-1-基)甲基)-5-(2,4-二氟苯基)四氢呋喃-3-基)甲醇(20mmol)加到50ml dcm中,再加入吡啶(7.9g,100mmol)和4-二甲氨基吡啶(dmap,0.73g,6mmol)。氮气保护,降温至0℃一次性加入4-甲基苯磺酰氯(tsc1,13.3g,70mmol)升至室温反应过夜,反应液用20ml饱和氯化铵溶液淬灭,再用10ml饱和碳酸氢钠和10ml饱和食盐水各洗涤一次,无水硫酸钠干燥,减压浓缩得到黄色油状物粗品,加入10ml异丙醇,50~60℃搅拌溶解,滴加10ml正己烷,降至室温析晶搅拌1.5h,抽滤,用10ml正己烷洗
涤,得到白色固体化合物。该化合物结构经核磁共振氢谱(1h nmr)和lc-ms鉴定,其数据如下:1h nmr(400mhz,dmso-d6)δ8.28(s,1h),7.79(d,j=8.3hz,2h),7.75(s,1h),7.49(d,j=8.1hz,2h),7.23(dtd,j=15.8,9.1,4.7hz,2h),6.95(td,j=8.5,2.4hz,1h),4.50(s,2h),3.96
–
3.82(m,3h),3.55(dd,j=8.8,7.0hz,1h),2.43(s,3h),2.42
–
2.35(m,1h),2.29(ddd,j=10.5,8.0,2.1hz,1h),1.98(dd,j=13.1,8.1hz,1h).m/z:450.30[m h]
,448.10[m-h]-。
[0060]
第二步:1-((3r,5r)-(5-((1h-1,2,4-三唑-1-基)甲基)-5-(2,4-二氟苯基)四氢呋喃-3-基)-n-(苯并[d][1,3]二氧戊环-5-基甲基)甲胺的制备(化合物31)
[0061]
50ml的圆底烧瓶中加入((3s,5r)-5-((1h-1,2,4-三唑-1-基)甲基)-5-(2,4-二氟苯基)四氢呋喃-3-基)4-甲基苯磺酸甲酯[中间体(8)](70mg,2.95mmol),k2co3(53mg,3.8mmol)和10ml的n-甲基吡咯烷酮(nmp),搅拌混匀,并滴加3,4-亚甲二氧基苯甲胺溶液(49mg,3.54mmol),在80℃的油浴下搅拌5h,tlc检测反应的进行,冷却后,用乙酸乙酯(10ml*3)萃取混合物三遍,并用饱和食盐水(10ml*3)洗涤有机层三遍,无水硫酸钠干燥,减压浓缩得粗品,硅胶柱纯化(洗脱剂:dcm:meoh=15:1),得71mg白色固体(收率为60%)。该化合物核磁共振氢谱(1h nmr)和lc-ms表征数据见表1。
[0062]
实施例5:1-((2r,4r)-4-((苯并[d][1,3]二氧戊环-5-基氧基)甲基)-2-(2,4-二氟苯基)四氢呋喃-2-基)甲基)-1h-1,2,4-三唑的制备(化合物32)
[0063]
将芝麻酚(30mg,1.97mmol)加入到装有8ml dmf的圆底烧瓶中,在冰浴的情况下,后缓慢加入nah(14mg,0.58mmol),搅拌0.5h,在加入((3s,5r)-5-((1h-1,2,4-三唑-1-基)甲基)-5-(2,4-二氟苯基)四氢呋喃-3-基)4-甲基苯磺酸甲酯的制备[中间体(8)](80mg,0.17mmol)。将混合物微波反应3h,微波反应器设定温度90℃,最大输出功率为100w。反应结束后,用乙酸乙酯(10ml*3)萃取混合物三遍,并用饱和食盐水(10ml*3)洗涤有机层三遍,无水硫酸钠干燥,减压浓缩得粗品,硅胶柱纯化(洗脱剂:ea:pe=2:1),得60mg无色油状物(收率为85%)。该化合物核磁共振氢谱(1h nmr)和lc-ms表征数据见表1。
[0064]
实施例6:本发明合成的氮唑类化合物具有抗真菌作用,其药理实验结果如下:
[0065]
(一)实验方法:采用常规的体外抑菌实验方法
[0066]
1.菌悬液配制:白色念珠菌(candida albicans)经yepd液体培养基35℃培养活化16小时,使真菌处于对数生长期后期,用血细胞计数板计数,以rpmi 1640液体培养基调整菌浓度至103个/ml。
[0067]
2.待测药液配制:取本发明待测化合物溶于二甲亚砜,配成6.4mg/ml的药物储存液,实验前用rpmi1640稀释成320μg/ml,并倍比稀释10个浓度。
[0068]
3.铺板及加药:96孔板1号孔加rpm1640 200μl作空白对照;3~12号孔各加菌悬液100μl,2号孔加菌悬液180μl和药液20μl。2~11号孔的药物浓度作10级倍比稀释,各孔药物终浓度依次为32、16、8、4、2、1、0.5、0.25、0.125、0.0625μg/ml。12号孔不加药液,作阳性对照,对照药物选用氟康唑。
[0069]
4.培养及检测:35℃培养箱孵育24小时,用酶标仪测定od
630
,设阳性对照孔光密度值(od值)为100%,以光密度值低于阳性对照孔50%的最低药物浓度为最小抑菌浓度值(mic)。mic高于最高浓度32μg/ml时,计为“>32μg/ml”,若mic为最低浓度或在最低浓度以下时,计为“mic≤0.0625μg/ml”,如若待测化合物的mic≤0.0625μg/ml,则采用各孔药物终浓度依次为4、2、1、0.5、0.25、0.125、0.0625、0.0312、0.0156、0.008μg/ml进一步测试。若
mic为最低浓度或在最低浓度以下时,不作区别,均计为“mic≤0.008μg/ml”。
[0070]
(二)实验结果
[0071]
化合物体外抗真菌实验结果见表2。
[0072]
表2 化合物体外抗真菌最小抑菌浓度(mic,μg/ml)
[0073][0074][0075]
上述实验结果表明,本发明的化合物大部分具有良好的抗真菌活性特点,部分化合物对白色念珠菌的抑制优于阳性药氟康唑,具有优秀的抗真菌活性。
[0076]
以上所述的仅是本发明的优选实施方式,应当指出,对于本领域的普通技术人员来说,在不脱离本发明创造构思的前提下,还可以做出若干变化和改进,这些都属于本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。