1.本发明涉及有机合成技术领域,具体涉及一种烯胺衍生物及其制备方法。
背景技术:
2.烯胺是一类含有双键、且双键上的碳原子与氮原子相连的不饱和胺类化合物,具有良好的稳定性和反应活性,其在医药、生物、有机化学合成等领域都具有广泛的应用,在一些天然的药物分子中存在烯胺结构,通过与吸电子基团或芳香基结合,降低其固有的亲和性;同时烯胺也可以作为各种官能团化反应的基础构型,进一步转化为烯酰胺或烯胺酮的烯胺衍生物,因此构筑多官能团化或者具有e/z导向性的烯胺衍生物成为了研究热点。
3.传统构筑烯胺衍生物的方法有缩合反应、加成反应、杂环裂解和使用亚胺制备烯胺等,而这些烯胺衍生物的制备方法存在明显的缺陷:(1)反应条件的要求较高,需要在高温高压或是极低温条件下才能反应);(2)对反应原料不同功能性基团的容忍度低、氨基试剂种类有限;(3)对反应体系要求高,部分反应需要保证反应体系无水无氧、催化剂种类少和用量高、需要特殊配体等;(4)反应原料的限制性,如原料类型单一、制备繁琐困难等。这些缺陷极大地限制了烯胺衍生物在医药、生物和化学领域的发展。
4.在硫代酰胺与α-重氮羰基化合物的金属催化偶联中,重氮化合物转化为金属卡宾,进而与硫代酰胺发生反应,生成叶立德,其通过环硫化物中间体导致烯胺酮的形成。第一例报道的金属卡宾与硫代酰胺反应是在1993年schulte等(参见kim g.,chu-moyer m.y.,danishefsky s.j.,et al.the total synthesis of indolizomycin[j].j.am.chem.soc.,1993,115(1):30-39)使用乙酸铑rh2(oac)4作为催化剂实现了分子内的偶联反应,但是其底物范围非常有限。因此,开发从易得的原料制备烯胺化合物的高效转化方法引起了人们的广泛关注,本发明所提供的新的利用锰催化剂制备烯胺化合物的方法具有重要意义。
技术实现要素:
[0005]
本发明的目的在于克服现有技术的不足之处而提供一种成本较低、制备方法简单、无需使用过多添加剂的烯胺衍生物的制备方法,且具有较高产率和扩大反应底物的选择性。
[0006]
为了实现上述目的,本发明采取的技术方案为:
[0007]
第一方面,本发明提供了一种烯胺衍生物的制备方法,包括如下步骤:将硫代酰胺化合物、重氮化合物、催化剂和有机溶剂混合,在惰性气体的条件下反应,得到所述烯胺衍生物;所述催化剂包括酞菁化锰;
[0008]
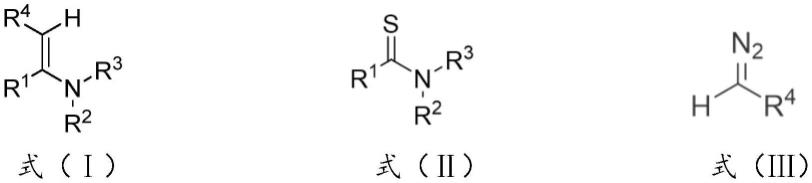
所述烯胺衍生物的结构式如下式(ⅰ)所示,所述硫代酰胺化合物的结构式如下式(ⅱ)所示,所述重氮化合物的结构式如下式(ⅲ)所示,
[0009][0010]
式(ⅰ)中,r1、r2、r3、r4分别选自烷基、环烷基、芳烷基、酯基、芳基中的至少一种,r2、r3为独立地取代基或相连形成环烷基;式(ⅱ)中,r1、r2、r3分别选自烷基、环烷基、芳烷基、酯基、芳基中的至少一种,r2、r3为独立地取代基或相连形成环烷基;式(ⅲ)中,r4选自烷基、环烷基、芳烷基、酯基、芳基中的至少一种。
[0011]
本发明所采用的催化剂酞菁化锰(mn(ⅱ)pc)和重氮化合物反应生成能够锰卡宾配合物,与本发明所述硫代酰胺化合物进一步反应生成硫叶立德中间体,金属缔合的叶立德中间体和无金属的叶立德中间体都可以进行电环化以形成环硫化物,随后在脱硫的同时会产生烯胺类的产物。
[0012]
本发明的发明人在大量的催化剂对比研究中发现,通过加入本发明所述酞菁化锰催化剂,能够高效地催化本发明所述硫代酰胺化合物与重氮化合物的偶联反应的进行。一方面,本发明无需使用昂贵、毒性较高的贵金属催化剂,进一步降低反应的成本;另一方面,使用其他的锰化物如氯化锰或羰基锰与本发明重氮化合物不能生成锰卡宾配合物,也不能与硫代酰胺化合物进一步发生反应,导致最终没有目标产物的生成。
[0013]
作为本发明所述烯胺衍生物的制备方法的优选实施方式,所述硫代酰胺化合物在有机溶剂中的物质的量浓度为0.05mol/l~0.2mol/l。
[0014]
本发明的发明人进行大量研究发现,将硫代酰胺化合物在有机溶剂中的物质的量浓度控制在上述范围内,本发明所述制备烯胺衍生物的产率较高,尤其是当硫代酰胺化合物在有机溶剂中的物质的量浓度优选为0.05mol/l~0.1mol/l时,所述烯胺衍生物的制备方法得到目标产物的产率能够达到84%以上。而随着硫代酰胺化合物在有机溶剂中的物质的量浓度增加时,会导致本发明制备烯胺衍生物的产率逐渐降低,在硫代酰胺化合物浓度较高时不利于本发明制备烯胺衍生物反应的进行。
[0015]
作为本发明所述烯胺衍生物的制备方法的优选实施方式,所述硫代酰胺化合物在有机溶剂中的物质的量浓度为0.05mol/l~0.1mol/l。
[0016]
作为本发明所述烯胺衍生物的制备方法的优选实施方式,所述硫代酰胺化合物、重氮化合物和催化剂的摩尔比为硫代酰胺化合物:重氮化合物:催化剂=1:(0.8~2):(0.001~0.05)。
[0017]
研究表明,当硫代酰胺化合物、重氮化合物和催化剂的摩尔比控制在上述范围内时,本发明所述烯胺衍生物的制备方法得到的目标产物具有较高的产率,有利于本发明硫代酰胺化合物和重氮化合物的交叉偶联反应的进行,而反应体系中催化剂的用量过多则会影响本发明制备烯胺衍生物的反应速率,导致目标产物的产率降低。
[0018]
作为本发明所述烯胺衍生物的制备方法的优选实施方式,所述硫代酰胺化合物、重氮化合物和催化剂的摩尔比为硫代酰胺化合物:重氮化合物:催化剂=1:1.8:(0.001~0.005)。
[0019]
本发明的发明人研究发现,当硫代酰胺化合物和重氮化合物的用量一定的条件
下,催化剂的用量降低至0.001~0.005时,仍然能保持较高的催化活性,制备得到的烯胺衍生物具有较高的产率,有利于降低反应成本。
[0020]
作为本发明所述烯胺衍生物的制备方法的优选实施方式,所述有机溶剂包括四氢呋喃(thf)、n,n-二甲基甲酰胺、二乙二醇二甲基醚、乙腈和叔丁醇中的至少一种。
[0021]
作为本发明所述烯胺衍生物的制备方法的优选实施方式,所述反应的温度为60℃~120℃,反应的时间为8h~48h。
[0022]
研究发现,当反应的温度和反应的时间控制在上述范围内时,有利于催化剂在反应过程中进行充分溶解,使得本发明所述烯胺衍生物的制备方法得到的目标产物具有较高的产率,有利于本发明硫代酰胺化合物和重氮化合物的交叉偶联反应的进行。而单一改变反应条件,将时间降低至12h以下或温度降低至80℃以下时,本发明所述烯胺衍生物的制备方法的产率为0,反应时间低于12h或反应温度低于80℃均会导致催化剂不能完全溶解,从而使得本发明所述烯胺衍生物的制备方法未能得到目标产物。
[0023]
作为本发明所述烯胺衍生物的制备方法的优选实施方式,所述反应的温度为80℃,反应的时间为16h。
[0024]
另一方面,本发明还提供了一种由烯胺衍生物的制备方法得到的烯胺衍生物。
[0025]
与现有技术相比,本发明的有益效果为:本发明通过采用过渡金属锰化物酞菁化锰作为催化剂,成本较低,原料易得,还能够有利于促进硫代酰胺化合物和重氮化合物的交叉偶联反应的进行,且本发明所述硫代酰胺化合物具有较广泛的底物选择范围。
附图说明
[0026]
图1为本发明实施例12的(e)-3-(二异丙基氨基)-3-苯基丙烯酸乙酯的1h-nmr谱图;
[0027]
图2为本发明实施例13的(e)-3-(二苯基氨基)-3-苯基丙烯酸乙酯的1h-nmr谱图;
[0028]
图3为本发明实施例14的(e)-3-(二丙基氨基)-3-苯基丙烯酸乙酯的1h-nmr谱图;
[0029]
图4为本发明实施例15的(e)-3-(二苄基氨基)-3-苯基丙烯酸乙酯的1h-nmr谱图;
[0030]
图5为本发明实施例16的(e)-3-(二苄基氨基)-3-(邻甲苯基)丙烯酸乙酯的1h-nmr谱图;
[0031]
图6为本发明实施例17的(e)-3-(4-氯苯基)-3-(二苄基氨基)丙烯酸乙酯的1h-nmr谱图;
[0032]
图7为本发明实施例18的(e)-3-(4-氰基苯基)-3-(二苄基氨基)丙烯酸乙酯的1h-nmr谱图;
[0033]
图8为本发明实施例19的(e)-3-(4-甲氧基苯基)-3-(二苄基氨基)丙烯酸乙酯的1h-nmr谱图;
[0034]
图9为本发明实施例20的(e)-3-(二苄基氨基)-1,3-二苯基丙-2-烯-1-酮的1h-nmr谱图。
具体实施方式
[0035]
下面结合实施例和附图,对本发明的技术方案作进一步说明。显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普
通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。实施例所使用的方法或操作,如无特别说明,均为本领域的常规方法或常规操作。
[0036]
一、反应条件的研究
[0037]
实施例1~11和对比例1~8
[0038]
本发明实施例1~11和对比例1~8的一种烯胺衍生物,本实施例1~11和对比例1~8所述烯胺衍生物为(e)-3-苯基-3-(1-吡咯烷基)丙烯酸酯,其结构式如下式(
ⅰ‑
a)所示,其制备方法的各组分及其用量和反应条件如下表1所示,
[0039][0040]
本发明实施例1~11和对比例1~8的制备烯胺衍生物所用的硫代酰胺化合物的结构式如式(
ⅱ‑
a)所示,其制备方法参见文献wei j,li y,jiang x.aqueous compatible protocol to both alkyl and aryl thioamide synthesis[j].organic letters,2016,47(22):340,具体的制备方法为:
[0041]
将升华硫s(320mg,10mmol)、对甲苯磺酸(43mg,0.25mmol)和苯甲醛(1.06g,10mmol)加入干净的双口瓶中,加入哌啶(20mmol),混合均匀,将双口瓶放置在120℃的油浴中回流搅拌12h。待反应冷却后,用二氯甲烷(20ml)溶解反应所得混合物,并将其转移至梨形瓶(50ml)中,随后用旋转蒸发仪减压浓缩除去溶剂,用硅胶柱层析纯化(洗脱剂为石油醚:乙酸乙酯=10:1,v/v),得到所述硫代酰胺化合物。
[0042]
本发明实施例1~11和对比例1~8的(e)-3-苯基-3-(1-吡咯烷基)丙烯酸酯的制备方法包括以下步骤:
[0043]
取干燥洁净的schlenk反应管,放入干净的磁子,拧上schlenk反应管塞,直至为半开状态,将其连接双排管,用热风枪加热并置换氮气5次。待schlenk反应管冷却至室温后,称量硫代酰胺化合物、催化剂、重氮乙酸乙酯和有机溶剂,置于schlenk反应管中,密封,在氮气条件下加热反应,得到粗产物。将粗产物用10ml二氯甲烷溶液洗涤,并加入中性氧化铝旋干,通过中性氧化铝柱层析纯化(洗脱剂为石油醚:乙酸乙酯=25/1,v/v),得到目标产物。
[0044]
表1
[0045]
[0046][0047]
将实施例1~11和对比例1~8制备得到的目标产物及其产率如下表2所示。
[0048]
表2
[0049]
[0050][0051]
由表1和表2可知,实施例1~11所述烯胺衍生物的产率都具有较高的产率,其产率均高于75%,其中实施例1制备得到的烯胺衍生物的产率最高,达到92%。
[0052]
比较实施例1与对比例1~2可知,本发明所述羰基锰作为催化剂能够与底物硫代酰胺化合物和重氮化合物发生反应,具有较高的目标产物的产率,且不是所有的锰化物都可以与本发明的硫代酰胺化合物和重氮化合物反应产生硫叶立德中间体,使用其他的锰化物如氯化锰或羰基锰,在其它各组分及用量均保持一致的条件下,对比例1~2均未能得到目标产物。
[0053]
比较实施例1~4与对比例3~4可知,本发明反应在特定的硫代酰胺化合物在有机溶剂中的物质的量浓度范围内,具有较高的目标产物的产率,其产率均达到84%以上,而当增加或降低硫代酰胺化合物在有机溶剂中的物质的量浓度时,均会导致目标产物的产率降低。
[0054]
比较实施例5~8与对比例5可知,当硫代酰胺化合物、重氮化合物和催化剂的摩尔比控制在1:(0.8~2):(0.001~0.05)时本发明反应制备得到的目标产物产率均能达到77%以上,特别是当硫代酰胺化合物和重氮化合物的用量比一定时,降低催化剂的用量时,本发明反应制备得到的目标产物的产率仍然能达到86%以上,有利于硫代酰胺化合物和重氮化合物的交叉偶联反应的进行,一方面降低催化剂的用量仍然能够保持较高的催化活
性,另一方面能够大大降低反应成本;而继续增加反应体系中催化剂的用量则会影响本发明制备烯胺衍生物的反应速率,导致目标产物的产率降低,当硫代酰胺化合物、重氮化合物和催化剂的摩尔比为1:1.8:0.1时,目标产物的产率仅有49%。
[0055]
比较实施例1、实施例9~11与对比例6~8可知,在本发明所述反应温度和反应时间的条件下,能够使得催化剂充分溶解,有利于硫代酰胺化合物和重氮化合物的交叉偶联反应的进行。而反应温度过高会使本发明所述制备烯胺衍生物的反应速率过快,从而导致目标产物的产率大大降低;若保证其他条件一致的情况下,反应时间低于12h或者反应温度低于80℃时,对比例7和对比例8均不能得到目标产物,反应无法进行。
[0056]
二、反应底物的研究
[0057]
实施例12
[0058]
本发明实施例的一种烯胺衍生物的制备方法,本实施例所述制备烯胺衍生物为(e)-3-(二异丙基氨基)-3-苯基丙烯酸乙酯,其反应机理如下所示,所述烯胺衍生物的化学式如式(
ⅰ‑
b)所示,
[0059][0060]
本实施例制备烯胺衍生物所用的硫代酰胺化合物的结构式如式(
ⅱ‑
b)所示,其制备方法参见文献wei j,li y,jiang x.aqueous compatible protocol to both alkyl and aryl thioamide synthesis[j].organic letters,2016,47(22):340,具体的制备方法为:
[0061]
(1)将二异丙基胺(1.01g,10mmol)、三乙胺(2.8ml,20mmol)和二氯甲烷(50ml)混合均匀,在0℃的条件下,加入苯甲酰氯(1.546g,11mmol),反应2h。反应完成后,用5ml饱和碳酸氢钠水溶液淬灭反应,用25ml二氯甲烷萃取三次,用25ml饱和nacl水溶液洗涤有机相三次,收集有机相,加入适量的无水na2so4干燥3小时,用砂芯漏斗抽滤过滤,用旋转蒸发仪减压浓缩除去溶剂,得到所述粗产物;
[0062]
(2)用25ml甲苯溶液溶解步骤(1)所述粗产物,加入劳森试剂(2.02g,5.0mmol),在120℃的条件下回流搅拌12h。反应完成后,待其冷却至室温后,用隔膜泵减压浓缩除去溶剂,用硅胶柱层析纯化(洗脱剂为石油醚:乙酸乙酯=10:1,v/v),得到所述硫代酰胺化合物。
[0063]
本实施例所述烯胺衍生物的制备方法与实施例1的制备方法相同,仅将实施例1中硫代酰胺化合物替换成同等物质的量的本实施例的硫代酰胺化合物。
[0064]
本实施例所得目标产物的产量为131.2mg,产率为95%,对目标产物进行1h-nmr表征,其1h-nmr的谱图如图1所示,表征如下:1h nmr(300mhz,cdcl3):δ7.42
–
7.33(m,3h),7.22
–
7.14(m,2h),4.98(s,1h),3.86(q,j=7.0hz,2h),3.73-3.60(m,2h),1.25(d,j=3.0hz,12h),1.05(t,j=6.0hz,3h).
13
c nmr(126mhz,cdcl3):δ168.0,160.7,138.6,128.3,128.0,128.0,87.9,58.3,20.7,14.6.hrms(esi)m/z:[m h]
calculated for c
17h26
o2n
:276.19581,found:276.19516,结构正确。
[0065]
实施例13
[0066]
本发明实施例的一种烯胺衍生物的制备方法,本实施例所述制备烯胺衍生物为(e)-3-(二苯基氨基)-3-苯基丙烯酸乙酯,其反应机理如下所示,所述烯胺衍生物的化学式如式(
ⅰ‑
c)所示,
[0067][0068]
本实施例制备烯胺衍生物所用的硫代酰胺化合物的结构式如式(
ⅱ‑
c)所示,其制备方法与实施例12的硫代酰胺化合物的制备方法相同,仅将步骤(1)中的二异丙基胺替换成同等物质的量的二苯胺。
[0069]
本实施例所述烯胺衍生物的制备方法与实施例1的制备方法相同,仅将实施例1中硫代酰胺化合物替换成同等物质的量的本实施例的硫代酰胺化合物。
[0070]
本实施例所得目标产物的产量为157.3mg,产率为92%,对目标产物进行1h-nmr表征,其1h-nmr的谱图如图2所示,表征如下:1h nmr(300mhz,cdcl3):δ7.43
–
7.37(m,2h),7.24
–
7.17(m,7h),7.08
–
7.01(m,6h),5.28(s,1h),3.95(q,j=7.0hz,2h),1.07(t,j=7.5hz,3h).
13
c nmr(101mhz,cdcl3):δ167.8,161.4,146.4,135.6,130.5,129.3,129.0,127.7,127.3,125.1,102.7,59.4,14.3.hrms(esi)m/z:[m h]
calculated for c
23h22
o2n
:344.16451,found:344.16382,结构正确。
[0071]
实施例14
[0072]
本发明实施例的一种烯胺衍生物的制备方法,本实施例所述制备烯胺衍生物为(e)-3-(二丙基氨基)-3-苯基丙烯酸乙酯,其反应机理如下所示,所述烯胺衍生物的化学式如式(
ⅰ‑
d)所示,
[0073][0074]
本实施例制备烯胺衍生物所用的硫代酰胺化合物的结构式如式(
ⅱ‑
d)所示,其制备方法与实施例12的硫代酰胺化合物的制备方法相同,仅将步骤(1)中的二异丙基胺替换成同等物质的量的二丙胺。
[0075]
本实施例所述烯胺衍生物的制备方法与实施例1的制备方法相同,仅将实施例1中硫代酰胺化合物替换成同等物质的量的本实施例的硫代酰胺化合物。
[0076]
本实施例所得目标产物的产量为119.8mg,产率为87%,对目标产物进行1h-nmr表征,其1h-nmr的谱图如图3所示,表征如下:1h nmr(300mhz,cdcl3):δ7.41
–
7.35(m,3h),7.20
–
7.16(m,2h),4.79(s,1h),3.88(q,j=7.0hz,2h),3.02(s,4h),1.61-1.53(m,4h),1.05(t,j=7.5hz,3h),0.84
–
0.78(m,6h).
13
c nmr(101mhz,cdcl3):δ168.2,162.6,137.2,
nmr(101mhz,cdcl3):δ167.8,162.2,136.1,135.8,130.0,128.8,128.6,128.3,127.5,126.0,87.8,58.6,19.7,14.5.hrms(esi)m/z:[m h]
calculated for c
26h28
o2n
:386.21146,found:386.21075,结构正确。
[0090]
实施例17
[0091]
本发明实施例的一种烯胺衍生物的制备方法,本实施例所述制备烯胺衍生物为(e)-3-(4-氯苯基)-3-(二苄基氨基)丙烯酸乙酯,其反应机理如下所示,所述烯胺衍生物的化学式如式(
ⅰ‑
g)所示,
[0092][0093]
本实施例制备烯胺衍生物所用的硫代酰胺化合物的结构式如式(
ⅱ‑
g)所示,其制备方法与实施例15的硫代酰胺化合物的制备方法相同,仅将步骤(1)中苯甲酰氯替换成同等物质的量的4-氯苯甲酰氯。
[0094]
本实施例所述烯胺衍生物的制备方法与实施例1的制备方法相同,仅将实施例1中硫代酰胺化合物替换成同等物质的量的本实施例的硫代酰胺化合物。
[0095]
本实施例所得目标产物的产量为192.8mg,产率为95%,对目标产物进行1h-nmr表征,其1h-nmr的谱图如图6所示,表征如下:1h nmr(300mhz,cdcl3):δ7.39
–
7.27(m,10h),7.15
–
7.12(m,4h),5.07(s,1h),4.30(s,4h),3.90(q,j=7.0hz,2h),1.07(t,j=7.5hz,3h).
13
c nmr(101mhz,cdcl3):δ167.9,161.9,136.7,135.0,134.8,130.0,128.9,128.8,127.6,127.2,89.7,58.9,52.7,14.5.hrms(esi)m/z:[m h]
calculated for c
25h25
o2ncl
:406.15683,found:406.15604,结构正确。
[0096]
实施例18
[0097]
本发明实施例的一种烯胺衍生物的制备方法,本实施例所述制备烯胺衍生物为(e)-3-(4-氰基苯基)-3-(二苄基氨基)丙烯酸乙酯,其反应机理如下所示,所述烯胺衍生物的化学式如式(
ⅰ‑
h)所示,
[0098][0099]
本实施例制备烯胺衍生物所用的硫代酰胺化合物的结构式如式(
ⅱ‑
h)所示,其制备方法与实施例15的硫代酰胺化合物的制备方法相同,仅将步骤(1)中苯甲酰氯替换成同等物质的量的4-氰基苯甲酰氯。
[0100]
本实施例所述烯胺衍生物的制备方法与实施例1的制备方法相同,仅将实施例1中硫代酰胺化合物替换成同等物质的量的本实施例的硫代酰胺化合物。
[0101]
本实施例所得目标产物的产量为163.2mg,产率为78%,对目标产物进行1h-nmr表征,其1h-nmr的谱图如图7所示,表征如下:1h nmr(300mhz,cdcl3):δ7.69
–
7.65(m,2h),7.47
–
7.43(m,2h),7.38
–
7.28(m,6h),7.14
–
7.12(m,2h),5.11(s,1h),4.29(s,4h),3.89(q,
j=7.0hz,2h),1.07(t,j=7.5hz,3h).
13
c nmr(101mhz,cdcl3):δ167.6,160.8,141.6,136.4,132.2,129.4,129.0,127.8,127.1,118.7,112.7,90.0,59.1,53.0,14.4.hrms(esi)m/z:[m h]
calculated for c
26h25
o2n
2
:397.19105,found:397.19031,结构正确。
[0102]
实施例19
[0103]
本发明实施例的一种烯胺衍生物的制备方法,本实施例所述制备烯胺衍生物为(e)-3-(4-甲氧基苯基)-3-(二苄基氨基)丙烯酸乙酯,其反应机理如下所示,所述烯胺衍生物的化学式如式(
ⅰ‑
i)所示,
[0104][0105]
本实施例制备烯胺衍生物所用的硫代酰胺化合物的结构式如式(
ⅱ‑
i)所示,其制备方法与实施例15的硫代酰胺化合物的制备方法相同,仅将步骤(1)中苯甲酰氯替换成同等物质的量的4-甲氧基苯甲酰氯。
[0106]
本实施例所述烯胺衍生物的制备方法与实施例1的制备方法相同,仅将实施例1中硫代酰胺化合物替换成同等物质的量的本实施例的硫代酰胺化合物。
[0107]
本实施例所得目标产物的产量为144.4mg,产率为72%,对目标产物进行1h-nmr表征,其1h-nmr的谱图如图8所示,表征如下:1h nmr(300mhz,cdcl3):δ7.37
–
7.23(m,8h),7.16
–
7.14(m,4h),6.95
–
6.90(m,2h),5.06(s,1h),4.30(s,4h),3.93(q,j=7.0hz,2h),3.81(s,3h),1.09(t,j=7.5hz,3h).
13
c nmr(101mhz,cdcl3):δ168.2,163.4,160.0,129.9,128.8,128.5,127.4,127.3,113.9,89.3,58.7,55.3,52.5,14.5.hrms(esi)m/z:[m h]
calculated for c
26h28
o3n
:402.20637,found:402.20560,结构正确。
[0108]
实施例20
[0109]
本发明实施例的一种烯胺衍生物的制备方法,本实施例所述制备烯胺衍生物为(e)-3-(二苄基氨基)-1,3-二苯基丙-2-烯-1-酮,其反应机理如下所示,所述烯胺衍生物的化学式如式(
ⅰ‑
j)所示,
[0110][0111]
本实施例制备烯胺衍生物所用的硫代酰胺化合物与实施例15的硫代酰胺化合物相同。本实施例制备烯胺衍生物所用的重氮化合物的结构式为式(
ⅲ‑
a),其具体的制备方法为:
[0112]
称量1,2-双(对甲苯基磺酰)肼(978.8mg,2.8mmol)和2-溴苯乙酮(521mg,2.6mmol),加入到烧瓶中,用氮气置换3次,加入四氢呋喃(15ml),搅拌均匀,在0℃条件下反应30min,加入dbu(593.7mg,3.9mmol),反应3h。完成反应后,加入5ml饱和碳酸氢钠水溶液淬灭反应,用20ml乙酸乙酯萃取,随后有机相用20ml饱和nacl水溶液洗涤三次,收集所有的
有机相,加入适量的无水硫酸镁干燥三小时后、用砂芯漏斗抽滤过滤,用旋转蒸发仪旋干,通过硅胶柱层析纯化(洗脱剂为石油醚:乙酸乙酯=10:1,v/v),得到目标产物。
[0113]
本实施例所述烯胺衍生物的制备方法与实施例15的制备方法相同,仅将实施例15中重氮乙酸乙酯替换成同等物质的量的本实施例的重氮化合物。
[0114]
本实施例所得目标产物的产量为181mg,产率为90%,对目标产物进行1h-nmr表征,其1h-nmr的谱图如图9所示,表征如下:1h nmr(300mhz,cdcl3):δ7.65(d,j=4.5hz,2h),7.40
–
7.23(m,18h),6.13(s,1h),4.42(s,4h).
13
c nmr(101mhz,cdcl3):δ188.0,163.8,141.6,136.7,130.9,129.0,128.8,128.8,128.7,128.4,128.0,127.8,127.7,127.3,96.5,53.2.hrms(esi)m/z:[m h]
calculated for c
29h26
on
:404.20089,found:404.20020,结构正确。
[0115]
最后所应当说明的是,以上是实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。