1.本发明属于中间体合成技术领域,具体涉及一种3-溴-6-异丙基-1氢-哌咯[2,3-b] 并吡啶的制备方法。
背景技术:
[0002]
近年来,针对癌症细胞的小分子靶向药物研发,是全球科研工作者最关心、科研投入最大的研究课题之一。这些替尼类小分子抗肿瘤药物的母核结构大多含有喹唑啉等骨架,属于吡啶类稠杂环。特别地,1h-哌咯[2,3-b]并吡啶及其衍生物作为潜在的5-羟色胺受体激动剂或部分激动剂,被广泛用于治疗阿兹海默病。而官能化的4-氮杂吲哚分子可作为糖原合成酶激酶-3的拮抗剂,为开发治疗糖尿病的药物领域提供了一种新的先导化合物。目前,多种吡啶类稠杂环结构的药物活性不断受到药物化学工作者的关注,但快速、高效地构建吡啶类稠杂环的合成方法,相关研究还处于萌芽状态。
[0003]
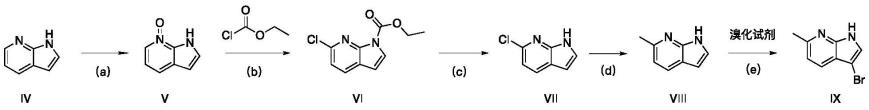
现有技术中1h-哌咯[2,3-b]并吡啶及其衍生物的合成路线具有复杂、合成成本高、不适宜工业生产等缺点,如3-溴-6-甲基-1氢-哌咯[2,3-b]并吡啶的合成方法,具体包括:
[0004][0005]
试剂和条件:(a)3-氯过氧苯甲酸,1,2-二氯乙烷,20℃,2h。
[0006]
(b)氯甲酸乙酯,双三甲基硅基胺基锂,四氢呋喃,惰性气体保护,20℃,2h。
[0007]
(c)氢氧化钠,甲醇溶液,20℃,4h。
[0008]
(d)1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物,碳酸钾,120℃,2h,微波反应。
[0009]
(e)nbs,盐酸或硫酸,四氢呋喃,20℃,2~48h。
[0010]
该方法以化合物iv(1氢-哌咯[2,3-b]并吡啶)作为起始原料,涉及到使用微波反应仪,腐蚀性化学原料硫酸或盐酸。反应过程中使用昂贵的催化剂1,1'-双(二苯膦基)二茂铁二氯化钯(ii)二氯甲烷复合物,并且还用到了微波仪,收率较低,不适宜大规模生产。
技术实现要素:
[0011]
为解决上述问题,本发明提出一种3-溴-6-异丙基-1氢-哌咯[2,3-b]并吡啶及其制备方法,所述3-溴-6-异丙基-1氢-哌咯[2,3-b]并吡啶具有潜在药物活性的吡啶绸杂环结构。所述制备方法解决现有技术合成步骤繁琐,反应试剂昂贵,收率较低,不适宜大规模生产的技术问题。该制备方法具有合成步骤简洁、原料便宜、操作方便不需要借助微波反应仪、反应总体易于控制、收率合适等优点。
[0012]
本发明是通过以下技术方案实现的:
[0013]
1.本发明的目的在于提供一种3-溴-6-异丙基-1氢-哌咯[2,3-b]并吡啶,其化学
结构式为:
[0014][0015]
本发明的另一目的在于提供一种3-溴-6-异丙基-1氢-哌咯[2,3-b]并吡啶的制备方法,所述制备方法以2-硝基哌咯(化合物ii)作为起始原料,在还原试剂(钯碳)催化下,先进行氢化反应;之后加入3-乙氧基-2,2-二甲基环丁酮和路易斯酸,反应得到化合物iii;将化合物iii与溴化试剂反应,得到所述3-溴-6-异丙基-1氢-哌咯[2,3-b]并吡啶(化合物i)。
[0016]
进一步地,所述制备方法的合成路线包括如下内容:
[0017][0018]
优选的,所述2-硝基哌咯(化合物ii)制备化合物iii的步骤中,2-硝基哌咯、3
‑ꢀ
乙氧基-2,2-二甲基环丁酮、路易斯酸的摩尔比范围为1:1:2;
[0019]
优选的,所述化合ii制备化合物iii的步骤中,先将2-硝基哌咯溶于ea中,然后添加还原试剂,预先反应3小时;此后向上述反应体系中添加3-乙氧基-2,2-二甲基环丁酮和路易斯酸进行反应,得到化合物iii。
[0020]
优选的,化合物ii制备化合物iii的步骤中,使用的有机溶剂种类为ea。
[0021]
优选的,化合物ii制备化合物iii的步骤中,所述路易斯酸为四氯化锑、四氯化锡和三氟化硼乙醚中一种或两种以上。
[0022]
优选的,化合物ii制备化合物iii的步骤中,反应温度为20℃~50℃。
[0023]
优选的,化合物iii制备化合物i的步骤中,所述溴化试剂为n-溴代丁二酰亚胺、溴化钾或溴素。
[0024]
优选的,化合物iii制备化合物i的步骤中,化合物iii与碘化试剂摩尔比范围为 1:0.95~1:2;反应温度为0℃~50℃。
[0025]
本发明的一种3-溴-6-异丙基-1氢-哌咯[2,3-b]并吡啶及其制备方法至少具有如下有益技术效果:
[0026]
本发明提供了一种简便有效的合成路线,合成得到了未见报道过的3-溴-6-异丙基-1 氢-哌咯[2,3-b]并吡啶,解决了原有合成技术的缺乏,反应条件温和,操作简便,可以实现实验室和工业生产的快速制备。
具体实施方式
[0027]
为了使本发明的目的、技术方案及优点更加清晰,以下结合实施例,对本发明进行进一步详细描述。应当理解,此处所描述的具体实施例仅用于解释本发明,并不用于限定本发明。
[0028]
相反,本发明涵盖任何由权利要求定义的在本发明的实质和范围上做的替代、修改、等效方法以及方案。进一步,为了使公众对本发明有更好的了解,在下文对本发明的细节描述中,详尽描述了一些特定的细节部分。对本领域技术人员来说没有这些细节部分的描述也可以完全理解本发明。
[0029]
下面的实施例起说明本发明的作用。在实施例中,除非另有说明,份数按重量份数计算,百分率按重量百分率计算,温度为摄氏度。按重量计算的分数和按体积计算的份数之间的关系与克和立方厘米之间的关系相同。
[0030]
实施例中涉及到的反应试剂的缩写如下所示:
[0031]
nbs:n-溴代丁二酰亚胺;
[0032]
tea:三乙胺;
[0033]
dipea:n,n-二异丙基乙胺;
[0034]
pe:石油醚;
[0035]
thf:四氢呋喃;
[0036]
mtbe:甲基叔丁基酸;
[0037]
dcm:二氯甲烷;
[0038]
ea:乙酸乙酯。
[0039]
meoh:甲醇。
[0040]
实施例1
[0041]
本实施例提出一种3-溴-6-异丙基-1氢-哌咯[2,3-b]并吡啶的制备方法,所述制备方法的合成路线包括:
[0042][0043]
其中,化合物ii制备化合物iii的具体内容包括:
[0044]
将化合物ii(11.20g,100mmol,1.0eq.)溶于150ml meoh中,室温20℃下,加入钯碳(2.00g,0.18eq.),置换氮气三次后,通入氢气反应2h后化合物ii反应完全,过滤,滤液旋蒸除去meoh后,补加150mlthf溶剂,加入3-乙氧基-2,2-二甲基环丁酮(14.20g, 100mmol,1.0eq.),三氟化硼乙醚(42.60g,300mmol,3.0eq.)后,室温20℃下反应6h,制砂柱层析纯化(正庚烷/ea洗脱)得化合物iii为14.20g,收率89%.化合物iii的1hnmr (400mhz,dmso-d6)δ(ppm)9.98(br,1h),7.89-7.87(d,1h),7.57-7.56(m,2h), 7.17-7.16(d,1h),3.24-3.21(m,1h),1.34-1.33(d,6h).
[0045]
化合物iii制备化合物i的具体内容包括:
[0046]
将化合物iii(10.60g,65.4mmo1,1.0eq.)溶于100ml dmf中,加入溴化钾(23.30g, 196mmol,3.0eq.),碳酸钾(27.09g,196mmol,3.0eq.),加完后30℃反应4h,过滤,有机相浓缩,制砂柱层析纯化(正庚烷/ea洗脱)得化合物i为白色固体13.48g,收率86%. 化合物i的1hnmr(400mhz,cdcl3)δ(ppm)7.67-7.66(d,1h),7.20-7.18(d,1h), 6.70(s,1h),3.22-3.20(m,1h),1.33-1.32(d,6h).(esi-tof)m/z:[m h]
calcdfor c10h11n2br:238;found:
239.纯度:98%。
[0047]
实施例2
[0048]
本实施例提出一种3-溴-6-异丙基-1氢-哌咯[2,3-b]并吡啶的制备方法,所述制备方法的合成路线包括:
[0049][0050]
其中,化合物ii制备化合物iii的具体内容包括:
[0051]
将化合物ii(11.20g,100mmol,1.0eq.)溶于150ml meoh中,室温20℃下,加入氢氧化钯/碳(2.00g,0.18eq.),置换氮气三次后,通入氢气反应6h后化合物ii反应完全,过滤,滤液旋蒸除去meoh后,补加150mlthf溶剂,加入3-乙氧基-2,2-二甲基环丁酮 (14.20g,100mmol,1.0eq.),四氯化钛(56.90g,300mmol,3.0eq.)后,室温20℃下反应3h,制砂柱层析纯化(正庚烷/ea洗脱)得化合物iii为13.60g,收率85%。化合物iii 的1hnmr(400mhz,dmso-d6)δ(ppm)9.98(br,1h),7.89-7.87(d,1h),7.57-7.56 (m,2h),7.17-7.16(d,1h),3.24-3.21(m,1h),1.34-1.33(d,6h).
[0052]
化合物iii制备化合物i的具体内容包括:
[0053]
将化合物iii(10.00g,62.5mmo1,1.0eq.)溶于100ml乙腈中,分批加入nbs(11.68g, 65.6mmol,1.05eq.),加完后室温20℃反应4h,过滤,滤饼用200ml水洗涤,将白色滤饼烘干得到化合物i白色固体14.50g,收率97%。化合物i的1hnmr(400mhz,cdcl3)δ(ppm) 7.67-7.66(d,1h),7.20-7.18(d,1h),6.70(s,1h),3.22-3.20(m,1h),1.33-1.32 (d,6h).(esi-tof)m/z:[m h]
calcd for c10h11n2br:238;found:239.纯度:98%。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
