可用于调节ahr信号传导的吡啶并嘧啶衍生物
1.本发明涉及如本文所述和定义的通式(i)化合物、用于制备所述化合物的方法、包括所述化合物的药物组合物和组合以及所述化合物和药物组合物的用途,所述化合物和药物组合物作为单独药剂或与其它活性成分组合,用于治疗或预防疾病,具体地癌症或免疫功能失调的病状或与异常ahr信号传导相关联的其它病状。此类化合物还可以用于扩增造血干细胞(hsc),以及在自体或同种异体移植中使用hsc来治疗患有遗传免疫疾病和自身免疫疾病以及各种造血病症的患者。
背景技术:
2.芳烃受体(ahr)是配体活化因子,其所属的家族为碱性螺旋-环-螺旋-per/arnt/sim家族。在配体在细胞质中结合后,ahr与其复合物以及hsp90和ahr相互作用蛋白xap2解离,从而使连接的ahr易位到细胞核。在细胞核中,ahr与ahr核转位子(arnt)二聚,然后与异型生物质应答元件(xre)结合,从而促进许多不同组织中的大量靶基因的上调或下调。ahr以与环境毒素结合并且诱导细胞色素p450家族的各个成员,包含消除其所需的cyp1a1、cyp1a2和cyp1b1而闻名。异型生物质对ahr的活化已经证明,此受体在一系列生理过程中发挥作用,所述生理过程包含胚胎发生、肿瘤发生和发炎(esser和rannug,《药物综述(pharmacol rev)》2015,67:259;roman等人,《药物疗法(pharmacol ther)》2018,185:50)。
3.ahr在包含树突状细胞、巨噬细胞、t细胞、nk细胞和b细胞的许多免疫细胞类型中表达并且在免疫调节中发挥重要作用(quintana和sherr,《药物综述》,2013,65:1148;nguyen等人,《免疫学前沿(front immunol)》,2014,5:551)。经典外源性ahr激动剂,如2,3,7,8-四氯二苯并-对-二噁英(tcdd)的毒性/不良作用是众所周知的并且包含深刻的免疫抑制和对恶性肿瘤的引发(esser等人,《免疫学趋势(trends immunol)》,2009,30:447;feng等人,《生物化学与生物物理学学报(biochimica et biophysica acta)》,2013,1836:197)。ahr激动剂对免疫细胞的生理作用包含促进调节性t细胞(treg)生成(pot,《瑞士医学周刊(swiss med wkly)》,2012,142:w13592)、调节th17细胞分化和活化(baricza等人,《细胞和分子生命科学(cell mol life sci)》,2016,73:95)和刺激白细胞介素-22(il-22)表达和/或从人活化的外周血单核细胞和t细胞中释放(ramirez等人,《欧洲免疫学杂志(eur j immunol)》,2010,40:2450;effner等人,《科学报告(sci rep)》,2017,7:44005)。ahr还调节抗原呈递细胞,如树突状细胞和巨噬细胞的功能。ahr活化会降低ii类主要组织相容性复合体和共刺激分子的表达并且还会降低通过树突状细胞进行的th1和th17极化细胞因子的产生(mezrich等人,《免疫学杂志(j immunol)》,2010,185:3190;nguyen等人,《美国国家科学院院刊(proc natl acad sci usa)》,2010,107:19961;quintana等人,2010《美国国家科学院院刊》,107:20768)。事实上,ahr活化会增强dc促进treg分化的能力(jurado-manzano等人,2017,《免疫学快报(immunol lett)》,190:84)。
4.除了异型生物质外,ahr还可以结合色氨酸降解的代谢产物,包含犬尿氨酸(kyn)和犬尿酸(kyna)。吲哚胺2,3双加氧酶1和2(ido1/ido2)以及色氨酸2,3-双加氧酶2(tdo2)催化kyn代谢途径的重要步骤并且在免疫细胞(ido1)和一系列癌细胞(ido1和tdo2)中表达
(pilotte等人,《美国国家科学院院刊》2012,109:2497)。作为刺激免疫系统识别和消除癌细胞的潜在新治疗,ido1的抑制剂已经吸引了广泛兴趣(cheong和sun,《药理科学趋势(trends pharmacol sci)》,2018,39:307)。传统上,ido1的免疫抑制作用主要归因于色氨酸的水平降低,所述色氨酸会活化激酶gcn2(一般性调控阻遏2)并且会抑制肿瘤引流淋巴结和肿瘤微环境两者中的t细胞增殖/活化。更最近地,很明显ido抑制剂的功效中的一些功效可能是ahr激动剂的产生减少的结果。这些内源产生的ahr激动剂已经示出了会引发对免疫细胞的一系列影响,包含上调树突状细胞中的ido1(julliard等人,《免疫学前沿》,2014,5:458)、抑制人t细胞增殖(frumento等人,《实验医学期刊(j exp med)》2002;196:459;terness等人,《实验医学期刊》,2002;196:447;opitz等人,《自然(nature)》,2011,478:197)以及上调细胞毒性t淋巴细胞中pd-1表达(liu等人,《癌症细胞(cancer cell)》,2018;33:480)。如上所突出的,ido1不是内源性ahr激动剂的唯一来源。tdo2主要在肝脏中表达,但其在一些癌症中也会组成性地表达,显著地在恶性神经胶质瘤、肝细胞癌、黑色素瘤、膀胱癌、乳腺癌、肺癌和结肠直肠癌(opitz等人,《自然(nature)》,2011,478:197;pilotte等人,《美国国家科学院院刊》,2012,109:2497;d
′
amato等人,《癌症研究(cancer res)》,2015,75(21):4651;hsu等人,《肿瘤靶标(oncotarget)》,2016,7(19):27584;chen等人,《疾病标志(dis markers)》,2016,2016:8169724)。此类数据表明,ahr拮抗剂可能比选择性ido-1抑制剂具有更广泛的功效,因为无论其来源如何,其都会减弱内源性ahr激动剂信号传导。最近发现的另一种酶即白细胞介素-4诱导1(il4i1)能够产生内源性ahr激动剂(sadik等人,《细胞(cell)》,2020,182:10),这一断言得到更多重视。
5.除了其对免疫细胞的影响之外,此类内源性激动剂还牵扯通过对肿瘤的直接影响的癌进展。例如,kyn增加人胶质母细胞瘤细胞存活和迁移(opitz等人,《自然》,2011,478:197)。几项其它研究也牵扯癌症进展中的环境配体不存在的情况下的ahr。ahr阻遏物(ahrr)蛋白在几种人类癌症中充当肿瘤抑制基因(zudaire等人,《临床研究杂志(j clin invest)》2008,118:640)。乳腺癌细胞中的ahr表达和“组成型”(内源性配体驱动的)活性与肿瘤侵袭性相关(schlezinger等人,《生物化学(biol chem)》,2006,387:1175;yang等人,《细胞生物化学杂志(j cell biochem)》,2008,104:402)并且控制与肿瘤侵袭相关联的基因的表达(yang等人,《癌基因(oncogene)》,2005,24:7869)。非恶性人乳腺上皮细胞中的异位ahr表达会诱导上皮到间充质的转化以及细胞生长速率的>50%增加(brooks和eltom,《癌症药靶研究最新进展(curr cancer drug targets)》,2011,11:654),并且ahr敲低诱导的基因在与到侵袭性较低的表型的间充质到上皮细胞逆转一致的人乳腺癌细胞系中会改变(narasimhan等人,《国际分子科学杂志(int j mol sci)》,2018,19:1388)。ahr拮抗剂或ahr敲低已经示出会降低人乳腺癌细胞在培养物中的增殖、存活、侵袭和迁移(parks等人,《分子药理学(molecular pharmacology)》2014,86:593;d
′
amato等人,《癌症研究(cancer res)》,2015,75(21):4651;narasimhan等人,《国际分子科学杂志》,2018,19:1388)并且会降低胶质母细胞瘤细胞的存活(gramatzki等人,《癌基因》,2009,28:2593;opitz等人,《自然》,2011,478:197;guastella等人,《神经肿瘤学杂志(j neuro-oncol),2018,印刷中)。最后,ahr拮抗剂阻断肿瘤球的形成(stanford等人,《分子癌症研究(mol cancer res)》,2016,14:696),所述肿瘤球是由癌症干细胞(csc)形成的,所述癌症干细胞是驱动肿瘤的启动、进展和转移的肿瘤细胞的子集。
6.因此,从免疫细胞和肿瘤细胞释放的ahr激动剂以自分泌和旁分泌方式起作用以促进肿瘤生长。因此,减少或阻断这些作用的药剂可用于治疗癌症和/或免疫功能失调的病状。因此,这类药剂还可用于一系列其它疾病/病状,包含但不限于肥胖症(rojas等人,《国际肥胖症杂志(int j obesity)》,2020,44:948)和各种病毒感染(giovannoni等人,《自然神经科学(nat neurosci.)》2020,23:939;giovannoni等人,《研究广场(res sq.)》2020,rs.3.rs-25639)。
7.wo2017/202816涉及用于治疗或预防癌症或免疫应答失调的病状或与异常ahr信号传导相关联的其它病症的化合物和组合物。具体地,wo2017/202816wo2018/146010和wo2019/101642尤其涉及能够抑制ahr功能的杂环化合物。
8.wo2020/081840涉及芳烃受体拮抗剂,如取代的咪唑并吡啶和咪唑并吡嗪,以及通过在这些试剂存在下培养造血干细胞或祖细胞来扩增造血干细胞的方法。
9.wo2020/039093涉及使用四氢吡啶并嘧啶衍生物作为ahr调节剂的组合物和方法。
10.wo2018/153893涉及6-酰胺基-1h-吲哚-2-基化合物,其可作为芳烃受体(ahr)调节剂,并且具体地作为ahr拮抗剂。本发明另外涉及化合物通过所述化合物结合所述芳烃受体来治疗和/或预防疾病和/或病状的用途。
11.wo2020/021024涉及双环化合物,其可作为芳烃受体(ahr)调节剂,并且具体地作为ahr拮抗剂。本发明另外涉及化合物通过所述化合物结合所述芳烃受体来治疗和/或预防疾病和/或病状的用途。
12.wo2020/043880涉及作为arh抑制剂的杂环化合物,其作为与其它活性成分组合的单独药剂,用于预防疾病,具体地癌症或免疫功能失调的病状或与异常ahr信号传导相关联的其它病状。
13.wo 2020/018848涉及至少部分通过使用拮抗ahr的化合物来扩增干细胞和/或谱系定向祖细胞,如造血干细胞和/或谱系定向祖细胞的方法。
14.wo2020/050409涉及具有芳烃受体拮抗剂活性并且可用于促进血小板生成的新型杂环化合物。
15.wo 2019/236766涉及至少部分通过使用拮抗ahr的内酰胺化合物来扩增干细胞和/或谱系定向祖细胞的方法。
16.wo2019/018562涉及使用杂芳基酰胺作为ahr调节剂化合物的组合物和方法,用于治疗至少部分由ahr调节的疾病。
17.wo 2018/195397涉及用于吲哚ahr抑制剂的组合物和方法。
18.wo 2018/146010涉及2-杂芳基-3-氧代-2,3-二氢哒嗪-4-甲酰胺的制备,其作为单独药剂或与其它活性成分组合,用于治疗或预防疾病,具体地癌症或免疫应答失调的病状。
19.wo2010/059401涉及用于扩增cd34 细胞的数量以用于移植的化合物和组合物。具体地,wo 2010/059401尤其涉及能够下调ahr的活性和/或表达的杂环化合物。
20.wo2012/015914涉及用于调节ahr活性的组合物和方法。具体地,wo2012/015914尤其涉及调节ahr活性以用于在治疗组合物中使用以抑制癌细胞增殖以及肿瘤细胞侵袭和转移的杂环化合物。
21.wo2020/051207涉及ahr拮抗剂以及通过在这些药剂存在下培养造血干细胞或祖
细胞来调节ahr活性和扩增造血干细胞的方法。此外,本公开提供通过施用这些ahr拮抗剂来治疗各种病变如癌症的方法
22.us2018/327411 a1涉及可用作ahr抑制剂以治疗与ahr相关联的多种疾病、病症和病状的化合物和组合物。
23.us2019/389857 a1涉及可作为ahr调节剂,并且具体地作为ahr拮抗剂的化合物。
技术实现要素:
24.本公开提供了通式(i)的嘧啶化合物,所述嘧啶化合物抑制ahr。本公开在以下段落中进行了概括:
25.1.一种式(i)化合物
[0026][0027]
其中:
[0028]
y为任选地包括1个、2个或3个选自n、o和s的杂原子的5元或6元环,所述5元或6元环被r5和r6取代;
[0029]
r1为h、c
1-3
烷基、(-ch2)pcn、-coc
1-3
烷基、-co(ch2)qnr7r8、-so2c
1-3
烷基、-so2nr7r8、-(ch2)qph、-c(o)z;
[0030]
r2为h或c
1-3
烷基;
[0031]
r3为h或c
1-3
烷基;
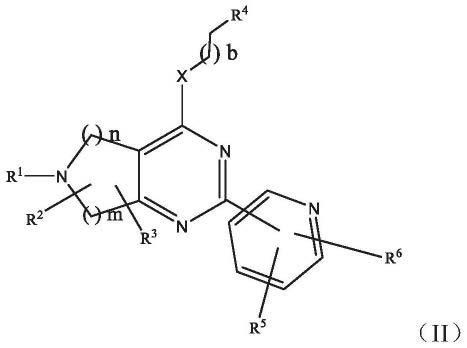
[0032]
r4为具有至少一个选自n、o或s的杂原子的9元或10元杂芳基(如吲哚-3-基或苯并咪唑-2-基),所述9元或10元杂芳基具有取代基r9和r
10
;
[0033]
r5为h、羟基、卤素(如f、cl)、cn、c
1-3
烷基、c
1-3
烷氧基(如ome)、c
1-2
卤代烷基(如cf3)、c
1-3
烷基(oh)、-co(ch2)qnr7r8、-so2c
1-3
烷基、-so2nr7r8,
[0034]
r6为h、羟基、卤素(如f、cl)、cn、c
1-3
烷基、c
1-3
烷氧基(如ome)、c
1-2
卤代烷基(如cf3)、c
1-3
烷基(oh)、-co(ch2)qnr7r8、-so2c
1-3
烷基、-so2nr7r8(如h、羟基、卤素(如f、cl)、cn、c
1-3
烷基、-co(ch2)qnr7r8、-so2c
1-3
烷基、-so2nr7r8),
[0035]
r7为h或c
1-3
烷基,如-ch3;
[0036]
r8为h或c
1-3
烷基,如-ch3;
[0037]
r9为h、羟基、卤素(如f、cl)、cn、c
1-3
烷基、c
1-3
烷氧基(如ome)、c
1-2
卤代烷基(如cf3)、c
1-3
烷基(oh)、-co(ch2)qnr7r8、-so2c
1-3
烷基或-so2nr7r8,
[0038]r10
为h、羟基、卤素(如f、cl)、cn、c
1-3
烷基、c
1-3
烷氧基(如ome)、c
1-2
卤代烷基(如cf3)、c
1-3
烷基(oh)、-co(ch2)qnr7r8、-so2c
1-3
烷基或-so2nr7r8(如h、羟基、卤素(如f、cl)、cn、c
1-3
烷基、-co(ch2)qnr7r8、-so2c
1-3
烷基或-so2nr7r8),
[0039]r11
为h或c
1-3
烷基(如-ch3);
[0040]
x为ch2、s、-so2、nr
11
或o;
[0041]
z为具有至少一个选自n、o和s的杂原子,例如1个或2个氮的5元或6元杂芳基,其中所述杂芳基任选地具有一个或两个选自羟基、卤素(如f、cl)、cn、c
1-3
烷基的取代基;
[0042]
b为0、1、2或3(例如0或2);
[0043]
n为整数1或2;
[0044]
m为整数1或2;
[0045]
p为整数1、2或3(如1);
[0046]
q为0、1、2或3(如0或1),
[0047]
或其药学上可接受的盐
[0048]
其条件是当x为nr
11
或o并且b为1或2时,那么r5或r9选自c
1-3
烷氧基(如ome)、c
1-2
卤代烷基(如cf3)和c
1-3
烷基(oh)。
[0049]
2.一种式(i)化合物,其中y为5元或6元含氮环。
[0050]
3.根据权利要求2所述的式(i)化合物,其中所述环为芳香族的。
[0051]
4.根据权利要求3所述的式(i)化合物,其中所述环为嘧啶或吡啶。
[0052]
5.根据权利要求4所述的式(i)化合物,其中r5位于5位。
[0053]
6.一种式(ii)化合物
[0054][0055]
其中x、r1、r2、r3、r4、r5、r6、b、m和n为以上针对式(i)化合物所定义的,或其药学上可接受的盐。
[0056]
7.一种式(iii)化合物:
[0057][0058]
其中x、r1、r2、r3、r4、r5、r6、b、m和n为以上针对式(i)化合物所定义的,或其药学上
可接受的盐。
[0059]
8.根据段落1至7中任一项所述的化合物,其中n为2。
[0060]
9.根据段落1至7中任一项所述的化合物,其中n为1。
[0061]
10.根据段落1至9中任一项所述的化合物,其中m为2。
[0062]
11.根据段落1至9中任一项所述的化合物,其中m为1。
[0063]
12.根据段落1至7中任一项所述的化合物,其具有式(iv):
[0064][0065]
其中x、r1、r2、r3、r4、r5、r6和b为以上针对式(i)化合物所定义的,或其药学上可接受的盐。
[0066]
13.根据段落1至7中任一项所述的化合物,其具有式(v):
[0067][0068]
其中x、r1、r2、r3、r4、r5、r6和b为以上针对式(i)化合物所定义的,或其药学上可接受的盐。
[0069]
14根据段落1至7中任一项所述的化合物,其具有式(vi):
[0070][0071]
其中x、r1、r2、r3、r4、r5、r6和b为以上针对式(i)化合物所定义的,或其药学上可接受的盐。
[0072]
15.根据段落1至7中任一项所述的化合物,其具有式(vii):
[0073][0074]
其中x、r1、r2、r3、r4、r5、r6和b为以上针对式(i)化合物所定义的,或其药学上可接受的盐。
[0075]
16.根据段落1至15中任一项所述的化合物,其中r1独立地选自h、ch3、-ch2ch3、-ch2ch2ch3、-ch(ch3)2、-c(o)ch3、c(o)nh2、-c(o)nhch3。-c(o)n(ch3)2、-ch2cn、-so2nh2、-so2ch3、-so2n(ch3)2、-ch2ph、-c(o)1-me-吡唑-5-基。
[0076]
17.根据段落1至15中任一项所述的化合物,其中r1独立地选自h、ch3、-ch2ch3、-ch2ch2ch3、-ch(ch3)2、-c(o)nh2、-c(o)nhch3。-c(o)n(ch3)2、-ch2cn、-so2nh2、-so2ch3、-so2n(ch3)2、-ch2ph、-c(o)1-me-吡唑-5-基。
[0077]
18.根据段落17所述的化合物,其中r1选自h、-ch2cn、-so2ch3和-so2n(ch3)2、-c(o)n(ch3)2。
[0078]
19.根据段落17或18所述的化合物,其中所述r1为h。
[0079]
20.根据段落1至15中任一项所述的化合物,其中r1为c
1-3
烷基,如-ch2ch3。
[0080]
21.根据段落1至20中任一项所述的化合物,其中r2为h或-ch3。
[0081]
22.根据权利要求21所述的化合物,其中r2为h。
[0082]
23.根据段落1至22中任一项所述的化合物,其中r3为h或-ch3。
[0083]
24.根据段落23所述的化合物,其中r3为h。
[0084]
25.根据段落1至24中任一项所述的化合物,其中r4选自各自独立地具有r9和r
10
的吲哚基(如吲哚-3-基,具体地5-氟-1h-吲哚-3-基)和苯并咪唑基(如苯并咪唑-2-基)。
[0085]
26.根据段落1至24中任一项的化合物,其中r5选自羟基、卤素(如f、cl)、cn、c
1-3
烷基、c
1-3
烷氧基(如ome)、c
1-2
卤代烷基(如cf3)、c
1-3
烷基(oh)、-co(ch2)qnr7r8、-so2c
1-3
烷基、-so2nr7r8[0086]
27.根据段落1至26中任一项所述的化合物,其中r5选自h、f、cl、-cf3、-so2ch3、-ch2ch2oh、cn、-och3和-ch3。
[0087]
28.根据段落1至25中任一项所述的化合物,其中r5为h。
[0088]
29.根据段落1至25中任一项所述的化合物,其中r5为f。
[0089]
30.根据段落1至25中任一项所述的化合物,其中r5为cl。
[0090]
31.根据段落1至25中任一项所述的化合物,其中r5为-cf3。
[0091]
32.根据段落1至25中任一项所述的化合物,其中r5为-so2ch3。
[0092]
33.根据段落1至25中任一项所述的化合物,其中r5为-ch2ch2oh。
[0093]
34.根据段落1至25中任一项所述的化合物,其中r5为cn。
[0094]
35.根据段落1至25中任一项所述的化合物,其中r5为-och3。
[0095]
36.根据段落1至25中任一项所述的化合物,其中r5为-ch3。
[0096]
37.根据段落1至36中任一项所述的化合物,其中r5为与分子的其余部分键合的原子的β。
[0097]
38.根据段落1至37中任一项所述的化合物,其中r6为h、f、cl、cn或-ch3。
[0098]
39.根据段落38所述的化合物,其中r6为h。
[0099]
40.根据段落1至39中任一项所述的化合物,其中r7选自h和-ch3。
[0100]
41.根据段落40所述的化合物,其中r7为-ch3。
[0101]
42.根据段落40所述的化合物,其中r7为h。
[0102]
43.根据段落1至42中任一项所述的化合物,其中r8选自h和-ch3。
[0103]
44.根据段落43所述的化合物,其中r8为h。
[0104]
45.根据段落43所述的化合物,其中r8为-ch3。
[0105]
46.根据段落1至45中任一项所述的化合物,其中r9选自h、f、cl、-cf3、-so2ch3、-ch2ch2oh、cn、-och3和-ch3。
[0106]
47.根据段落46所述的化合物,其中r9为h。
[0107]
48.根据段落46所述的化合物,其中r9为f。
[0108]
49.根据段落46所述的化合物,其中r9为cl。
[0109]
50.根据段落46所述的化合物,其中r9为cf3。
[0110]
51.根据段落46所述的化合物,其中r9为-so2ch3。
[0111]
52.根据段落46所述的化合物,其中r9为-ch2ch2oh。
[0112]
53.根据段落46所述的化合物,其中r9为cn。
[0113]
54.根据段落46所述的化合物,其中r9为-och3。
[0114]
55.根据段落46所述的化合物,其中r9为-ch3。
[0115]
56.根据段落1至55中任一项所述的化合物,其中r
10
为h。
[0116]
57.根据段落1至56中任一项所述的化合物,其中r
10
为甲基。
[0117]
58.根据段落1至57中任一项所述的化合物,其中r
11
为h。
[0118]
59.根据段落1至58中任一项所述的化合物,其中b为0。
[0119]
60.根据段落1至58中任一项所述的化合物,其中b为1。
[0120]
61.根据段落1至58中任一项所述的化合物,其中b为2。
[0121]
62.根据段落1至58中任一项所述的化合物,其中b为3。
[0122]
63.根据段落1至62中任一项所述的化合物,其中p为1。
[0123]
64.根据段落1至63中任一项所述的化合物,其中q为1。
[0124]
65.根据段落1至63中任一项所述的化合物,其中q为0。
[0125]
66.根据段落1至65中任一项所述的化合物,其中x为ch2[0126]
67.根据段落1至65中任一项所述的化合物,其中x为s。
[0127]
68.根据段落1至65中任一项所述的化合物,其中x为nr
11
。
[0128]
69.根据段落1至65中任一项所述的化合物,其中x为o。
[0129]
70.一种药物组合物,其包括根据段落1至69中任一项所述的化合物以及赋形剂、稀释剂或载体。
[0130]
71.一种根据权利要求1至69中任一项所述的化合物或一种根据段落70所述的药物组合物,其用于治疗。
[0131]
72.一种用于根据段落71使用的化合物或组合物,其用于治疗癌症。
[0132]
73.一种治疗患者的方法,所述方法包括施用治疗有效量的根据段落1至69中任一项所述的化合物或根据段落70所述的组合物。
[0133]
74.一种根据段落1至69中任一项所述的化合物或根据段落70所述的组合物的用途,其用于制造用于治疗癌症的药品。
[0134]
在一个实施例中,m为1,并且n为1。在一个实施例中,m为1,并且n为2。在一个实施例中,m为2,并且n为1。在一个实施例中,m为2,并且n为2。在一个实施例中,n为1,并且m为1或2。
[0135]
在一个实施例中,r4为吲哚,例如根据本公开被取代,如在选自2;2和5;和6的位置处被取代。
[0136]
在一个实施例中,r4为2-三氟甲基-1h吲哚-3-基。
[0137]
在一个实施例中,r4为6-甲氧基-1h吲哚-3-基。
[0138]
在一个实施例中,r4为5-甲氧基、2-甲基-1h吲哚-3-基。
[0139]
在一个实施例中,y为嘧啶,包含被r5和r6取代的嘧啶,例如其中r5或r6中的至少一个不为h。
[0140]
在一个实施例中,z是未经取代的。
[0141]
在一个实施例中,x为o或nh,y为吡啶基或嘧啶基,r1为h或乙基,r2为h,r3为h,r4为吲哚基(如吲哚-3基,具体地在2、2 5和6位被取代),r5为f、-och3或cf3(例如在5位),r6为h,r9为h、f和och3,包含其药学上可接受的盐。
[0142]
本公开还包含本文公开的个别化合物、其药用盐、其药物调配物和其中任一种的治疗用途,具体地如本文别处所述的。
[0143]
在一个实施例中,本公开提供实例1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17和18,其中任一种的药用盐和/或其中任一种的药物调配物。
[0144]
在一个实施例中,盐选自氯化物和盐酸盐。
[0145]
在一个实施例中,本公开提供:
[0146]
2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-4-胺);2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h-吡咯并[3,4-d]嘧啶-4-胺;n-[2-(1h-吲哚-3-基)乙基]-2-(5-甲氧基吡啶-3-基)-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-4-胺);n-[2-(1h-吲哚-3-基)乙基]-2-[5-(三氟甲基)吡啶-3-基]-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-4-胺);2-(5-氟吡啶-3-基)-4-{[2-(5-甲氧基-2-甲基-1h-吲哚-3-基)乙基]氨基}-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-鎓[如氯化物];2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h,9h-嘧啶并[4,5-d]氮杂卓-4-胺;7-乙基-2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h,9h-嘧啶并[4,5-d]氮杂卓-4-胺;3-(2-{[2-(5-氟吡啶-3-基)-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-4-基]氧基}乙基)-6-甲氧基-1h-吲哚[如盐酸盐];2-(5-氟吡啶-3-基)-4-({2-[2-(三氟甲基)-1h-吲哚-3-基]乙基}氨基)-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-鎓[如氯化物];其中任一种的药用盐和/或其中任一种的药物调配物。
[0147]
本公开的化合物扩展到包括原子的形式,这些原子是元素的更常见形式的“稀有”同位素,例如氘。
[0148]
在一个实施例中,本公开的化合物为ahr抑制剂。
[0149]
本公开的化合物包含其放射性标记形式。
[0150]
具体地,令人惊讶地发现本发明的化合物可有效抑制ahr。所述化合物可用于治疗或预防其中外源性和内源性ahr配体诱导免疫应答失调的病状,例如:不受控的细胞生长、肿瘤细胞的增殖和/或存活、免疫抑制。此失调可以在癌症、不适当的细胞免疫应答和不适当的细胞炎症应答的上下文中观察到。
[0151]
在一个实施例中,本公开的化合物可用于治疗癌症(例如液体和/或实体瘤)和/或其转移。癌症的实例包含头颈癌(如脑瘤和脑转移)、胸癌(包含非小细胞和小细胞肺癌)、胃肠癌(包含胃、食管、结肠和结直肠)、胆道癌、胰腺癌、肝癌、内分泌癌、乳腺癌、卵巢癌、膀胱癌、肾癌、前列腺癌、骨癌和皮肤癌。
[0152]
在一个实施例中,癌症是上皮癌。在一个实施例中,癌症是肉瘤。在一个实施例中,癌症是转移性的。
具体实施方式
[0153]
如任选地包括1个、2个或3个选自氮、氧和硫的杂原子的5元或6元环是指含有5个或6个原子的饱和、部分饱和或芳香族环,包含其中所有原子均为碳或其中存在1个、2个或3个独立地选自氮、氧和硫的杂原子,例如环戊二烯、苯基、噻吩、呋喃、吡咯、吡唑、咪唑、噁唑、噻唑、异噻唑、三唑、吡啶、吡嗪、三嗪、噻嗪、噁嗪、环戊烷、环己烷、吡咯烷、吡咯啉、吡唑烷、咪唑啉、四氢呋喃、四氢噻吩、哌啶、哌嗪、四氢吡喃、噻烷、噻喃、吗啉或硫代吗啉。
[0154]
在一个实施例中,环是5元的。
[0155]
在一个实施例中,环是6元的。
[0156]
在一个实施例中,5元或6元环是不饱和的或芳香族的(即5元或6元杂芳基)。
[0157]
如本文采用的5元或6元杂芳基是含有5个或6个原子的环,其中至少一个原子是杂
原子,例如选自氮、氧或硫的杂原子,如吡咯、吡唑、咪唑、噻吩、噁唑、异噻唑、噻唑、吡啶、哒嗪、吡嗪、三嗪、噻喃、噁嗪和噻嗪,如吡咯、吡唑和吡啶以及嘧啶。
[0158]
在一个实施例中,5元或6元环选自环戊二烯、苯基、吡啶和吡嗪,如苯基和吡啶。
[0159]
因此,在一个实施例中,y为吡啶或嘧啶(即包含被r5和r6取代)。
[0160]
在一个实施例中,y为吡唑。
[0161]
在一个实施例中,y不为吡唑。
[0162]
如本文所采用的c
1-3
烷基是指直链或支链烷基,例如甲基、乙基、丙基或异丙基。
[0163]
如本文所采用的卤素包含氟、氯、溴或碘。
[0164]
co代表羰基。
[0165]
如本文所采用的9元或10元杂芳基是指含有9个或10个原子的双环系统,其中至少一个环为芳香族,并且至少一个环含有杂原子,例如含有1个、2个、3个或4个独立地选自氮、氧和硫的杂原子,如二氢吲哚、吲哚、异吲哚、吲哚嗪、吲唑、苯并咪唑、氮杂吲哚、吡唑并嘧啶、嘌呤、苯并呋喃、异苯并呋喃、苯并噻吩、苯并异噁唑、苯并异噻唑、苯并噁唑、苯并噻二唑、腺嘌呤、鸟嘌呤、四氢喹啉、二氢异喹啉、喹啉、异喹啉、喹嗪、喹喔啉、酞嗪、噌啉、萘啶、吡啶并嘧啶、吡啶并吡嗪、吡啶并吡嗪、蝶啶、色烯、异色烯、色酮、苯并噁嗪、喹啉酮和异喹啉酮。
[0166]
在一个实施例中,9元或10元杂芳基选自吲哚基和苯并咪唑基,如吲哚-3-基或苯并咪唑-2-基。
[0167]
本公开的化合物可以通过本文描述的方法制备。
[0168]
可以采用一般途径1来制备本公开的某些化合物:
[0169][0170]
其中
[0171]
l1和l2为离去基团,例如卤素,如氯;
[0172]
l3为离去基团,例如硼酸;
[0173]
p1为保护基团,例如boc;
[0174]
x为nr
11
、o或s;和
[0175]
r4和y为以上针对式(i)化合物所定义的。
[0176]
可以采用一般途径2来制备本公开的某些化合物:
[0177][0178]
l1和l2为离去基团,例如卤素,如氯;
[0179]
l3为离去基团,例如硼酸;
[0180]
l4为离去基团,例如卤素,如溴;
[0181]
p1为保护基团,例如boc;
[0182]
x为nr
11
、o或s;和
[0183]
r1、r4和y为以上针对式(i)化合物所定义的。
[0184]
其中x为ch2的式(i)化合物可通过钯催化反应制备。
[0185]
位阻碱的实例是三乙胺,其在方案1的步骤1和方案2的步骤3中可以与色胺一起使用。
[0186]
方案1的步骤2中的合适缓冲剂是例如于如二噁烷和水等溶剂中的芳基硼酸和碳酸钾。
[0187]
偶联剂可能需要在氮气下执行反应。用于方案1的步骤2和方案2的步骤4中的合适偶联剂包含双(二苯基膦基)二茂铁]二氯化钯(ii)二氯。
[0188]
可以使用例如tfa,具体地于二氯甲烷中进行方案1的步骤3和方案2的步骤1中的脱保护。
[0189]
可以在位阻有机碱,如三乙胺的存在下进行方案2的步骤2。用于反应的合适的极性非质子溶剂是二氯甲烷。
[0190]
在上述一个或多个反应期间,可能需要保护基团来保护化学敏感性基团,以确保过程有效。因此,如果需要或必要,可以通过使用常规保护基团来保护中间体化合物。保护基团和用于去除所述保护基团的方法描述于theodora w.greene和peter g.m.wuts的“有机合成中的保护基团(protective groups in organic synthesis)”,约翰威利父子出版公司(john wiley&sons inc);第4修订版,2006,isbn-10:0471697540。
[0191]
本公开的化合物的盐的实例包含所有药学上可接受的盐,如但不限于强矿物酸的
酸加成盐,如hcl和hbr盐,以及强有机酸的加成盐,例如甲磺酸盐。
[0192]
本公开扩展到本文公开的化合物的溶剂化物。溶剂化物的实例包含水合物。
[0193]
新型中间体也是本发明的方面。
[0194]
本公开的另外的方面为制备本文公开的化合物和/或中间体的方法。
[0195]
本文还提供了药物组合物,所述药物组合物包括根据本公开的化合物和赋形剂、稀释剂或载体。对药学上可接受的载体的透彻讨论可见于《雷明顿氏药物科学(remington
′
s pharmaceutical sciences)》(新泽西州马克出版公司(mack publishing company,n.j.),1991)。
[0196]
本公开的药物组合物可以通过任何数量的途径施用,包含但不限于口服、静脉内、肌肉内、动脉内、髓内、鞘内、心室内、透皮、经皮(例如,参见wo98/20734)、皮下、腹膜内、鼻内、肠内、局部、舌下、阴道内或直肠途径。也可以使用无针注射器来施用本发明的药物组合物。
[0197]
在一个实施例中,治疗组合物可以以注射剂形式制备,无论是液体溶液还是悬浮液形式。也可以制备适于在注射之前溶解或悬浮在液体媒剂中的固体形式。用于重组此类固体形式(包含冻干的固体)的合适液体可以选自水性溶液,例如盐水、右旋糖或注射用水等。在一个实施例中,重组的液体调配物是等渗的。
[0198]
在一个实施例中,根据本公开的药物组合物以用于口服施用的片剂或胶囊剂的形式提供。
[0199]
治疗
[0200]
本公开还扩展到治疗患者的方法,所述方法包括施用例如用于治疗癌症的治疗有效量的本公开的化合物(或包括所述化合物的药物组合物)。
[0201]
还提供了一种用于治疗,例如用于治疗癌症的根据本公开的化合物(或包括所述化合物的药物组合物)。
[0202]
在另外的方面,提供了一种用于制造用于治疗癌症的药品的本公开的化合物(或包括所述化合物的药物组合物)。
[0203]
在一个实施例中,癌症是上皮癌,例如选自选自以下的实例肝癌(如肝细胞癌)、胆道癌、乳腺癌(如无er 乳腺癌)、前列腺癌、结肠直肠癌、卵巢癌、宫颈癌、肺癌、胃癌、胰腺癌、骨癌、膀胱癌、头颈癌、甲状腺癌、皮肤癌、肾癌和食道癌(例如胃癌)。
[0204]
在一个实施例中,癌症选自选自包括以下的组:肝细胞癌、胆道癌、乳腺癌、前列腺癌、结肠直肠癌、卵巢癌、肺癌、胃癌、胰腺癌和食道癌。
[0205]
在一个实施例中,胆管癌的位置选自肝内胆管、左肝管、右肝管、肝总管、胆囊管、胆总管、肝胰管壶腹和其组合。
[0206]
在一个实施例中,胆管癌在肝内胆管中。在一个实施例中,胆管癌在左肝管中。在一个实施例中,胆管癌在右肝管中。在一个实施例中,胆管癌在肝总管中。在一个实施例中,胆管癌在胆囊管中。在一个实施例中,胆管癌在胆总管中。在一个实施例中,胆管癌在乏特氏壶腹中。在一个实施例中,上皮癌是癌。
[0207]
在一个实施例中,根据本公开的治疗是辅助疗法,如外科手术之后。
[0208]
在一个实施例中,根据本公开的疗法是新辅助治疗,例如用于在外科手术之前缩小肿瘤。
[0209]
在一个实施例中,肿瘤是实体瘤。在一个实施例中,所述癌症是原发性癌症、继发性癌症、转移或其组合。在一个实施例中,根据本公开的治疗适合于继发性肿瘤的治疗。在一个实施例中,所述癌症是转移性癌症。在一个实施例中,根据本公开的治疗适合于治疗原发性癌症和转移。在一个实施例中,根据本公开的治疗适合于治疗继发性癌症和转移。在一个实施例中,根据本公开的治疗适合于治疗原发性癌症、继发性癌症和转移。
[0210]
在一个实施例中,根据本公开的治疗适合于治疗淋巴结中的癌细胞。
[0211]
在一个实施例中,肝癌是原发性肝癌。在一个实施例中,肝癌是继发性肝癌。在一个实施例中,肝癌是阶段1、2、3a、3b、3c、4a或4b。
[0212]
在一个实施例中,胃癌是0期、i期、ii期、iii期或iv期。
[0213]
对人类受试者的精确治疗有效量将取决于疾病状态的严重度、受试者的一般健康、受试者的年龄、体重和性别、饮食、施用的时间和频率、一种或多种药物组合、反应敏感性和对疗法的耐受性/应答。此量可以通过常规实验确定,并且在临床医生的判断内。通常,治疗有效量将为0.01mg/kg到1000mg/kg,例如0.1mg/kg到500mg/kg。药物组合物可以方便地以单位剂量形式呈现,所述单位剂量形式每剂量含有预定量的本发明的活性剂。
[0214]
组合疗法
[0215]
在一个实施例中,本公开的化合物用于例如其中另外的疗法是抗癌疗法的组合疗法。
[0216]
在一个实施例中,抗癌疗法是化学疗法。
[0217]
化学治疗剂和化学疗法或细胞毒性剂在本文中可互换使用,除非上下文另外指出。
[0218]
本文所采用的化学疗法旨在是指对恶性细胞和组织“选择性”破坏的特异性抗肿瘤化学药剂或药物,例如烷化剂、抗代谢物(包含胸苷酸合酶抑制剂)、蒽环类药物、抗微管剂(包含植物生物碱)、拓扑异构酶抑制剂、parp抑制剂和其它抗肿瘤剂。在此上下文中,选择性地使用是不严格的,因为当然这些药剂中的许多均具有严重的副作用。
[0219]
优选的剂量可以由医师基于所治疗的癌症的性质来选择。
[0220]
本公开的方法中可以采用的烷化剂的实例包含:烷化剂,所述烷化剂选自:氮芥、亚硝基脲、四嗪、氮丙啶、铂类和衍生物;以及非经典烷化剂。
[0221]
含有铂的化学治疗剂(也被称为铂类)包含例如顺铂、卡铂、奥沙利铂、沙铂、吡铂、奈达铂、三铂(triplatin)和脂铂(lipoplatin)(顺铂的脂质体形式),具体地顺铂、卡铂和奥沙利铂。
[0222]
顺铂的剂量范围为约20到约270mg/m2,这取决于确切癌症。剂量通常处于约70到约100mg/m2的范围内。
[0223]
氮芥包含二氯甲基二乙胺、环磷酰胺、美法仑、苯丁酸氮芥、异环磷酰胺和白消安。
[0224]
亚硝基脲包含n-亚硝基-n-甲基脲(mnu)、卡莫司汀(bcnu)、洛莫司汀(ccnu)和司莫司汀(meccnu)、福莫司汀和链脲霉素。四嗪包含达卡巴嗪(dacarbazine)、米托唑胺(mitozolomide)和替莫唑胺(temozolomide)。
[0225]
氮丙啶包含噻替派、丝裂霉素和亚丝醌(azq)。
[0226]
本公开的方法中可以采用的抗代谢药的实例包含抗叶酸剂(例如,氨甲蝶呤(methotrexate)和培美曲塞(pemetrexed))、嘌呤类似物(例如,硫嘌呤(thiopurine),如硫
唑嘌呤(azathiopurine)、巯嘌呤(mercaptopurine)、硫嘌呤、氟达拉滨(fiudarabine)(包含磷酸盐形式)、喷司他丁(pentostatin)和克拉屈滨(cladribine))、嘧啶类似物(例如,氟嘧啶(fluoropyrimidine),如5-氟尿嘧啶(fluorouracil)和其前药,如卡培他滨(capecitabine)氟尿苷(floxuridine)、吉西他滨(gemcitabine)、阿糖孢苷(cytarabine)、地西他滨(decitabine)、雷替曲塞(raltitrexed)(拓优得(tomudex))盐酸盐、克拉屈滨和6-氮尿嘧啶。
[0227]
本公开的方法中可以采用的蒽环类药物的实例包含柔红霉素(daunorubicin)(道诺霉素(daunomycin))、柔红霉素(脂质体)、多柔比星(doxorubicin)(亚德里亚霉素(adriamycin))、多柔比星(脂质体)、表柔比星(epirubicin)、伊达比星(idarubicin)、戊柔比星(valrubicin)(目前仅用于治疗膀胱癌)和米托蒽醌(mitoxantrone)、蒽环类类似物,具体地多柔比星。
[0228]
本公开的方法中可以采用的抗微管剂的实例包含包含长春花生物碱和紫杉烷。
[0229]
长春花生物碱包含完全天然的化学品,例如长春新碱和长春碱并且还包含半合成长春花生物碱,例如长春瑞滨、长春地辛和长春氟宁。
[0230]
紫杉烷包含紫杉醇、多西他赛、白蛋白结合型紫杉醇、卡巴他赛(carbazitaxel)和其衍生物。本文所采用的紫杉烷的衍生物包含如紫杉醇(taxol)等紫杉烷的再调配物,例如在胶束调配物中,衍生物还包含其中采用合成化学来修饰作为紫杉烷的起始材料的化学衍生物。
[0231]
本公开的方法中可以采用的拓扑异构酶抑制剂包含i型拓扑异构酶抑制剂、ii型拓扑异构酶抑制剂和ii型拓扑异构酶毒剂。i型抑制剂包含拓朴替康(topotecan)、伊立替康(irinotecan)、吲哚替康(indotecan)和因帝米替康(indimitecan)。ii型抑制剂包含染料木素(genistein)和icrf 193,其具有以下结构:
[0232][0233]
ii型毒剂包含安吖啶(amsacrine)、依托泊苷(etoposide)、磷酸依托泊苷(etoposide phosphate)、替尼泊苷(teniposide)和多柔比星以及氟喹诺酮(fluoroquinolone)。
[0234]
在一个实施例中,所采用的化学治疗剂的组合是例如铂和5-fu或其前药,例如顺铂或奥沙利铂以及卡培他滨或吉西他滨,如folfox。
[0235]
在一个实施例中,化学疗法包括化学治疗剂,具体地细胞毒性化学治疗剂的组合。
[0236]
在一个实施例中,化学疗法组合包括铂,如顺铂和氟尿嘧啶或卡培他滨。
[0237]
在一个实施例中,化学疗法组合包括卡培他滨和奥沙利铂(xelox)。
[0238]
在一个实施例中,化学疗法是亚叶酸和5-fu的组合,任选地与奥沙利铂组合。
[0239]
在一个实施例中,化学疗法是亚叶酸、5-fu和伊立替康(folfiri)的组合,任选地
与奥沙利铂(folfirinox)组合。所述方案由以下组成:伊立替康(180mg/m
2 iv,经历90分钟)与亚叶酸(400mg/m2[或2
×
250mg/m2]iv,经历120分钟)同时施用;随后是氟尿嘧啶(400-500mg/m
2 iv团注),然后是氟尿嘧啶(2400-3000mg/m2静脉内输注,经历46小时)。此周期通常每两周重复一次。上文所示的剂量可能因周期而异。
[0240]
在一个实施例中,化学疗法组合采用微管抑制剂,例如硫酸长春新碱、埃博霉素a、n-[2-[(4-羟苯基)氨基]-3-吡啶基]-4-甲氧基苯磺酰胺(abt-751)、紫杉醇衍生的化学治疗剂,例如太平洋紫杉醇、白蛋白结合型紫杉醇或多西他赛或其组合。
[0241]
在一个实施例中,化学疗法组合包括抗代谢药,如卡培他滨(希罗达(xeloda))、磷酸氟达拉滨、氟达拉滨(氟达拉(fludara))、地西他滨、雷替曲塞(拓优得(tomudex))、盐酸吉西他滨和克拉屈滨。
[0242]
在一个实施例中,抗癌症疗法组合采用mtor抑制剂。mtor抑制剂的实例包含:依维莫司(everolimus)(rad001)、wye-354、ku-0063794、雷帕霉素(papamycin)(西罗莫司(sirolimus))、替西罗莫司(temsirolimus)、地磷莫司(deforolimus)(mk-8669)、azd8055和bez235(nvp-bez235)。
[0243]
在一个实施例中抗癌症疗法组合采用mek抑制剂。mek抑制剂的实例包含:as703026、ci-1040(pd184352)、azd6244(司美替尼(selumetinib))、pd318088、pd0325901、azd8330、pd98059、u0126-etoh、bix 02189或bix 02188。
[0244]
在一个实施例中,化学疗法组合采用akt抑制剂。akt抑制剂的实例包含:mk-2206和at7867。
[0245]
在一个实施例中,抗癌症疗法采用极光激酶抑制剂。极光激酶抑制剂的实例包含:极光a抑制剂i、vx-680、azd1152-hqpa(巴拉塞替(barasertib))、sns-314甲磺酸、pha-680632、zm-447439、cct129202和橙皮苷(hesperadin)。
[0246]
在一个实施例中,化学疗法组合采用例如wo2010/038086中公开的p38抑制剂,如n-[4-({4-[3-(3-叔丁基-1-p-甲苯基-1h-吡唑-5-基)脲基]萘乙酰胺-1-基氧基}甲基)吡啶-2-基]-2-甲氧基乙酰胺基。
[0247]
在一个实施例中,组合采用bcl-2抑制剂。bcl-2抑制剂的实例包含:奥巴克拉甲磺酸酯(obatoclax mesylate)、abt-737、abt-263(纳威托克斯(navitoclax))和tw-37。
[0248]
在一个实施例中,化学疗法组合包括更昔洛韦(ganciclovir),其可以协助控制免疫应答和/或肿瘤血管形成。
[0249]
在一个实施例中,抗癌症疗法包含parp抑制剂。
[0250]
在一个实施例中,抗癌症疗法包含具有对dhodh酶的活性的特异性抑制的癌症代谢抑制剂。
[0251]
在一个实施例中,本文方法中采用的一个或多个疗法是有节奏的,即低剂量的抗癌药物的连续或频繁治疗,通常条件是伴随有其它疗法方法。
[0252]
在一个实施例中,提供了使用多个周期的治疗(如化学疗法),例如2个、3个、4个、5个、6个、7个或8个。
[0253]
在一个实施例中,本公开的化合物不是:
[0254]
吡啶并(3,4-d)嘧啶-4-胺,n-[2-(1h-苯并咪唑-2-基)乙基]-5,6,7,8-四氢-2-(4-吡啶基)cas登记号1331980-89-2;
[0255]
吡啶并(3,4-d)嘧啶-4-胺,n-[2-(1h-苯并咪唑-2-基)乙基]-5,6,7,8-四氢-2-(3-吡啶基)cas登记号1332109-12-2;
[0256]
吡啶并(3,4-d)嘧啶-4-胺,5,6,7,8-四氢-n-[2-(1h-吲哚-3-基)乙基]-2-(3-吡啶基)cas登记号1332134-49-2;
[0257]
5h-吡咯并(3,4-d)嘧啶-4-胺,6,7-二氢-n-(2-(1h-吲哚-3-基)乙基)-2-(3-吡啶基)cas登记号1360232-40-1;
[0258]
5h-吡咯并(3,4-d)嘧啶-4-胺,6,7-二氢-n-(2-(1h-吲哚-1-基)乙基)-2-(3-吡啶基)cas登记号;
[0259]
吡啶并(3,4-d)嘧啶-4-胺,5,6,7,8-四氢-n-[2-(1h-吲哚-3-基)乙基]-2-苯基cas登记号1360364-34-6;
[0260]
吡啶并(3,4-d)嘧啶-4-胺,5,6,7,8-四氢-n-[2-(1h-吲哚-3-基)乙基]-2-)2-吡啶基)cas登记号1360407-66-4。
[0261]“包括”在本说明书的上下文中旨在意指“包含”。在技术上适当的情况下,可以组合本发明的实施例。
[0262]
本文将实施例描述为包括某些特征/要素。本公开还扩展到分开由所述特征/要素组成或基本由所述特征/要素组成的实施例。
[0263]
如专利和申请等技术参考文献通过引用并入本文。
[0264]
本文具体地且明确地叙述的任何实施例可以单独或与一个或多个另外的实施例组合形成免责声明的基础。
[0265]
本技术要求于2020年2月26日提交的并且通过引用并入本文的sg10202001722x的优先权。此应用可以用作进行更正的基础。
[0266]
现将参考以下实例来描述本发明,这些实例仅是说明性的并且不应以被解释为限制本发明的范围。
[0267]
实例
[0268]
通用方法a(色胺)
[0269]
向合适的圆底烧瓶或反应小瓶装入芳基卤(1当量)、色胺(1.1当量)、ipa(10ml/mmol)和三乙胺(2当量),并且在100℃下加热,持续3小时(反应通过uplc分析监测)。一旦冷却,就将反应混合物蒸发至干燥,在乙酸乙酯与水之间分配所得残余物。分离有机相,并依序用饱和碳酸氢盐溶液、水、盐水进行洗涤,然后经硫酸钠干燥,过滤并蒸发。如果期望,通过色谱或研磨执行纯化。
[0270]
通用方法b(铃木(suzuki))
[0271]
向合适的圆底烧瓶或反应小瓶装入芳基卤(1当量)、芳基硼酸(1.5-2.0当量)、碳酸钾(1.5-2.0当量)、二噁烷/水([5:1]约60体积)。用氮气对项部空间进行冲洗,然后添加[1,1
′‑
双(二苯基膦基)二茂铁]二氯化钯(ii)二氯(0.2-0.3当量)。在氮气下将反应混合物于100℃下加热2-24小时,直到如通过uplc分析确定的完成为止。将反应混合物蒸发至干燥,并且以于dcm中的浆液形式施加到硅胶柱上;或者将其预先吸附到硅藻土上,然后将其装载到干式装载单元中并且与硅胶盒串联放置。用一定梯度的乙酸乙酯/己烷对所期望的产物进行洗脱,有时可能需要极性更大的甲醇(0-10%)/乙酸乙酯洗脱液。可能需要在用含7m氨的甲醇(0-10%)/dcm洗脱的硅胶上进行进一步色谱。用二乙醚研磨,并且随后进行过
滤,以得到所期望的产物。
[0272]
通用方法c(tfa deboc)
[0273]
向boc化合物(20-200mg)于dcm(3-10ml)的溶液中添加tfa(0.2-0.5ml)。一旦如通过uplc判断的完成,就将反应混合物装载到scx树脂盒(0.5g或1.0g)上。用甲醇(10ml)对盒进行彻底洗涤。将产物以游离碱形式进行洗脱,用含7m氨的甲醇(10ml)进行洗脱。对游离碱物质进行蒸发,用乙醚进行研磨,并且通过过滤收集。在《10mbar的干燥器中进行干燥。
[0274]
在2020年2月26日提交的优先权文件sg10202001722x中公开的参考样品1至12在wo2020/039093中公开,并且通过引用具体并入本文。
[0275]
表1中的以下实例通过与本文给出的方法类似的方法制备并且用于参考化合物1至12:
[0276]
[0277]
[0278][0279]
实例1 2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-4-胺)的制备
[0280][0281]
步骤1 2-氯-4-{[2-(5-甲氧基-1h-吲哚-3-基)乙基]氨基}-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯)
[0282]
根据通用方法a,使用2,4-二氯-5,6-二氢吡啶并[3,4-d]嘧啶-7(8h)羧酸叔丁酯(500mg)和2-(6-甲氧基-1h-吲哚-3-基)乙-1-胺(340mg)制备,以在通过由mtbe(10ml)研磨进行纯化后得到呈灰白色固体状的期望的产物(510mg,68%)
[0283]
uplc-ms(碱性方法,2分钟):rt 1.19分钟,m/z=458/460[m h] 氯同位素模式
[0284]
1h nmr(400mhz,dmso)δ10.74-10.54(m,1h),7.53(s,1h),7.23(d,j=8.7hz,1h),7.14(d,j=2.4hz,1h),7.05(d,j=2.4hz,1h),6.72(dd,j=8.8,2.4hz,1h),4.27(s,2h),3.76(s,3h),3.67-3.50(m,4h),2.92(t,j=7.6hz,2h),2.34(t,j=5.7hz,2h),1.43(s,9h)。
[0285]
步骤2 2-(5-氟吡啶-3-基)-4-{[2-(5-甲氧基-1h-吲哚-3-基)乙基]氨基}-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯)
[0286]
根据通用方法b,使用2-氯-4-{[2-(5-甲氧基-1h-吲哚-3-基)乙基]氨基}-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯)(500mg)和5-氟吡啶-3-硼酸(231mg)制备,以得到呈灰白色固体状的期望的产物(226mg,21%)。
[0287]
uplc-ms(碱性方法,2分钟):rt 1.27分钟,m/z 519.3[m h]
[0288]
19f nmr(400mhz,dmso-d6)δppm-127.61质子去耦
[0289]
1h nmr(400mhz,dmso)δ10.66(s,1h),9.32(d,j=1.7hz,1h),8.67(d,j=2.9hz,1h),8.42-8.19(m,1h),7.27(s,1h),7.22(d,j=8.7hz,1h),7.17(d,j=2.3hz,1h),7.00(d,j=2.4hz,1h),6.71(dd,j=8.8,2.4hz,1h),4.40(s,2h),3.79(q,j=6.8hz,2h),3.70(s,3h),3.68-3.57(m,2h),3.03(t,j=7.5hz,2h),2.44(t,j=5.9hz,2h),1.45(s,9h)。
[0290]
步骤3 2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-4-胺)
[0291]
根据通用方法c,使用2-(5-氟吡啶-3-基)-4-{[2-(5-甲氧基-1h-吲哚-3-基)乙基]氨基}-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯)(300mg)制备,以在通过制备型hplc纯化(碱性条件)进行纯化后得到呈白色固体状的期望的产物(54mg,23%)。
[0292]
uplc-ms(4分钟碱性):rt=1.57分钟,m/z=419.3[m h]
[0293]
19
f nmr(376mhz,dmso)δ-127.82质子去耦
[0294]1h nmr(400mhz,dmso)δ10.62(s,1h),9.31(d,j=1.7hz,1h),8.66(d,j=2.8hz,1h),8.36-8.24(m,1h),7.47(d,j=8.6hz,1h),7.15-7.01(m,2h),6.85(d,j=2.2hz,1h),6.64(dd,j=8.6,2.3hz,1h),3.82-3.73(m,5h),3.71(s,2h),2.99(t,j=5.1hz,4h),2.34(t,2h)。
[0295]
实例5 2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h-吡咯并[3,4-d]嘧啶-4-胺的制备
[0296][0297]
步骤1 2-氯-4-{[2-(6-甲氧基-1h-吲哚-3-基)乙基]氨基}-5h,6h,7h-吡咯并[3,4-d]嘧啶-δ-羧酸叔丁酯
[0298]
根据通用方法a,使用2,4-二氯-5h,6h,7h-吡咯并[3,4-d]嘧啶-6-羧酸叔丁酯(500mg)和2-(6-甲氧基-1h-吲哚-3-基)乙-1-胺(361mg)制备,以在通过由mtbe研磨进行纯化后得到呈黄色固体状的期望的产物(889mg,98%)
[0299]
uplc-ms(碱性方法,2分钟):rt 1.17分钟,m/z 444.3/446.3[m h] 氯同位素模式
[0300]
1h nmr(400mhz,dmso)δ10.62(s,1h),7.97(d,j=5.4hz,1h),7.49(dd,j=8.8,2.4hz,1h),7.03(d,j=2.2hz,1h),6.85(d,j=2.3hz,1h),6.65(dd,j=8.6,2.3hz,1h),4.46-4.25(m,4h),3.76(s,3h),3.58(q,j=7.0hz,2h),2.90(t,j=7.6hz,2h),1.46(d,j=4.3hz,9h)
[0301]
步骤2 2-(5-氟吡啶-3-基)-4-{[2-(6-甲氧基-1h-吲哚-3-基)乙基]氨基}-5h,6h,7h-吡咯并[3,4-d]嘧啶-6-羧酸叔丁酯
[0302]
根据通用方法b,使用2-氯-4-{[2-(6-甲氧基-1h-吲哚-3-基)乙基]氨基}-5h,6h,7h-吡咯并[3,4-d]嘧啶-6-羧酸叔丁酯(889mg)和5-氟吡啶-3-硼酸(565mg)制备,以得到呈白色固体状的期望的产物(333mg,33%)。
[0303]
uplc-ms(碱性方法,2分钟):rt 1.22分钟,m/z 505.4[m h]
[0304]
19f nmr(400mhz,dmso-d6)δppm-127.58质子去耦
[0305]
1h nmr(400mhz,dmso)δ10.61(d,j=2.2hz,1h),9.31(q,j=1.8hz,1h),8.69(d,j=2.9hz,1h),8.31(ddd,j=11.7,6.1,3.3hz,1h),7.71(d,j=4.9hz,1h),7.45(dd,j=8.6,2.2hz,1h),7.06(d,j=2.2hz,1h),6.84(d,j=2.2hz,1h),6.64(dd,j=8.6,2.3hz,1h),4.45(dd,j=19.9,12.6hz,4h),3.75(s,5h),2.98(t,j=7.4hz,2h),1.48(d,j=4.4hz,9h)
[0306]
步骤3 2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h-吡咯并[3,4-d]嘧啶-4-胺
[0307]
根据通用方法c,使用2-(5-氟吡啶-3-基)-4-{[2-(6-甲氧基-1h-吲哚-3-基)乙基]氨基}-5h,6h,7h-吡咯并[3,4-d]嘧啶-6-羧酸叔丁酯(333mg)制备,以在由mtbe研磨后得到呈白色固体状的期望的产物(120mg,45%)。
[0308]
uplc-ms(4分钟碱性):rt=1.40分钟,m/z=405.3[m h]
[0309]
19f nmr(376mhz,dmso)δ-127.73(s)
[0310]
1h nmr(400mhz,dmso)δ10.66(d,j=2.3hz,1h),9.37(t,j=1.7hz,1h),8.71(d,j=2.9hz,1h),8.38(ddd,j=10.1,2.9,1.6hz,1h),7.50(d,j=8.6hz,1h),7.46(t,j=5.8hz,1h),7.10(d,j=2.2hz,1h),6.89(d,j=2.3hz,1h),6.68(dd,j=8.6,2.3hz,1h),4.03(dd,j=7.0,2.1hz,4h),3.80(s,5h),3.03(dd,j=8.6,6.4hz,2h)
[0311]
实例9 n-[2-(1h-吲哚-3-基)乙基]-2-(5-甲氧基吡啶-3-基)-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-4-胺)的制备
[0312][0313]
步骤1 2-氯-4-{[2-(1h-吲哚-3-基)乙基]氨基}-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯)
[0314]
根据通用方法a,使用2,4-二氯-5,6-二氢吡啶并[3,4-d]嘧啶-7(8h)羧酸叔丁酯(500mg)和色胺(333mg)制备,以在通过由mtbe(10ml)研磨进行纯化后得到呈白色固体状的期望的产物(345mg,49%)
[0315]
uplc-ms(碱性方法,2分钟):rt 1.23分钟,m/z 428.3/430.3[m h] 氯同位素模式
[0316]
h nmr(400mhz,dmso)δ10.81(s,1h),7.64(d,j=7.8hz,1h),7.53(s,1h),7.34(d,j=8.0hz,1h),7.18(d,j=2.3hz,1h),7.06(td,j=8.1,7.0,1.2hz,1h),6.98(td,j=7.4,6.9,1.0hz,1h),4.27(s,2h),3.59(p,j=5.9,5.3hz,4h),2.95(t,j=7.6hz,2h),2.35(t,j=5.9hz,2h),1.42(s,9h)。
[0317]
步骤2 4-{[2-(1h-吲哚-3-基)乙基]氨基}-2-(5-甲氧基吡啶-3-基)-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯)
[0318]
根据通用方法b,使用2-氯-4-{[2-(1h-吲哚-3-基)乙基]氨基}-5h,6h,7h,8h-吡
啶并[3,4-d]嘧啶-7-羧酸1叔丁酯)(100mg)和(5-甲氧基吡啶-3-基)硼酸)(54mg)制备,以得到呈白色固体状的期望的产物(58mg,25%)。
[0319]
uplc-ms(碱性方法,2分钟):rt 1.21分钟,m/z 501.4[m h]
[0320]
1h nmr(400mhz,dmso)δ10.84(s,1h),9.09(d,j=1.6hz,1h),8.38(d,j=2.9hz,1h),8.11(dd,j=3.0,1.7hz,1h),7.59(d,j=7.8hz,1h),7.35(d,j=8.1hz,1h),7.26(s,1h),7.22(d,j=2.1hz,1h),7.07(t,j=7.4hz,1h),6.96(t,j=7.5hz,1h),4.41(s,2h),3.87(s,3h),3.80(q,j=7.0hz,2h),3.65(s,2h),3.05(t,j=7.6hz,2h),2.45(d,j=5.8hz,2h),1.45(s,9h)。
[0321]
步骤3 n-[2-(1h-吲哚-3-基)乙基]-2-(5-甲氧基吡啶-3-基)-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-4-胺)
[0322]
根据通用方法c,使用4-{[2-(1h-吲哚-3-基)乙基]氨基}-2-(5-甲氧基吡啶-3-基)-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯)(43mg)制备,以得到呈米色固体状的期望的产物(23mg,50%)。
[0323]
uplc-ms(4分钟碱性):rt=1.52分钟,m/z=401.3[m h]
[0324]
1h nmr(400mhz,dmso-d6)δ10.82(s,1h),9.08(d,j=1.6hz,1h),8.36(d,j=2.9hz,1h),8.10(dd,j=3.0,1.6hz,1h),7.60(d,j=7.8hz,1h),7.38-7.31(m,1h),7.21(d,j=2.2hz,1h),7.11-7.03(m,2h),6.97(ddd,j=8.0,7.0,1.0hz,1h),3.87(s,3h),3.79(d,j=10.9hz,4h),3.05(d,j=7.1hz,4h),2.37(s,2h)。缺少一个可交换质子
[0325]
实例10 n-[2-(1h-吲哚-3-基)乙基]-2-[5-(三氟甲基)吡啶-3-基]-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-4-胺)的制备
[0326][0327]
步骤1 2-氯-4-{[2-(1h-吲哚-3-基)乙基]氨基}-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯)
[0328]
根据通用方法a,使用2,4-二氯-5,6-二氢吡啶并[3,4-d]嘧啶-7(8h)羧酸叔丁酯(500mg)和色胺(333mg)制备,以在通过由mtbe(10ml)研磨进行纯化后得到呈白色固体状的
期望的产物(345mg,49%)
[0329]
uplc-ms(碱性方法,2分钟):rt 1.23分钟,m/z 428.3/430.3[m h] 氯同位素模式
[0330]
h nmr(400mhz,dmso)δ10.81(s,1h),7.64(d,j=7.8hz,1h),7.53(s,1h),7.34(d,j=8.0hz,1h),7.18(d,j=2.3hz,1h),7.06(td,j=8.1,7.0,1.2hz,1h),6.98(td,j=7.4,6.9,1.0hz,1h),4.27(s,2h),3.59(p,j=5.9,5.3hz,4h),2.95(t,j=7.6hz,2h),2.35(t,j=5.9hz,2h),1.42(s,9h)。
[0331]
步骤2 4-{[2-(1h-吲哚-3-基)乙基]氨基}-2-[5-(三氟甲基)吡啶-3-基]-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯)
[0332]
根据通用方法b,使用2-氯-4-{[2-(1h-吲哚-3-基)乙基]氨基}-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯)(100mg)和3-(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)-5-(三氟甲基)吡啶)(96mg)制备,以得到呈白色固体状的期望的产物(53mg,42%)。
[0333]
uplc-ms(碱性方法,2分钟):rt 1.35分钟,m/z 539.3[m h]
[0334]
19f nmr(400mhz,dmso-d6)δppm-60.94质子去耦
[0335]
1h nmr(400mhz,dmso)δ10.84(s,1h),9.71(d,j=1.9hz,1h),9.09(d,j=2.2hz,1h),8.84(s,1h),7.58(d,j=8.0hz,1h),7.35(s,1h),7.35-7.32(m,1h),7.21(d,j=2.2hz,1h),7.10-7.03(m,1h),7.00-6.93(m,1h),4.43(s,2h),3.79(d,j=9.6hz,2h),3.65(s,2h),3.05(t,j=7.7hz,2h),2.46(s,2h),1.45(s,9h)。
[0336]
步骤3 n-[2-(1h-吲哚-3-基)乙基]-2-[5-(三氟甲基)吡啶-3-基]-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-4-胺)
[0337]
根据通用方法c,使用4-{[2-(1h-吲哚-3-基)乙基]氨基}-2-[5-(三氟甲基)吡啶-3-基]-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯)(52mg)制备,以得到呈残余物状的期望的产物(26mg,61%)。
[0338]
uplc-ms(4分钟碱性):rt=1.82分钟,m/z=439.3[m h]
[0339]
19
f nmr(376mhz,dmso)δ-60.97质子去耦
[0340]1h nmr(400mhz,dmso)δ10.84(s,1h),9.70(d,j=1.9hz,1h),9.07(dd,j=2.3,1.0hz,1h),8.83(q,j=3.2,2.3hz,1h),7.60(d,j=7.8hz,1h),7.38-7.29(m,2h),7.20(d,j=2.2hz,1h),7.07(td,j=8.0,7.0,1.1hz,1h),6.97(td,j=8.0,7.0,1.1hz,1h),3.90(s,2h),3.80(q,j=7.4,7.0hz,2h),3.17(s,2h),3.05(t,j=7.7hz,2h),2.47(s,2h)。(1h缺少,nh)
[0341]
实例14 2-(5-氟吡啶-3-基)-4-{[2-(5-甲氧基-2-甲基-1h-吲哚-3
‑‑
基)乙基]氨基}-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-鎓氯化物的制备
[0342][0343]
步骤1 2-氯-4-{[2-(5-甲氧基-2-甲基-1h-吲哚-3-基)乙基]氨基}-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯
[0344]
根据通用方法a,使用2,4-二氯-5,6-二氢吡啶并[3,4-d]嘧啶-7(8h)羧酸叔丁酯(500mg)和2-(5-甲氧基-2-甲基-1h-吲哚-3-基)乙-1-胺(401mg)制备,以在使用biotage(telos柱10g,洗脱液己烷-etoac,5至30至50%)进行纯化后得到呈褐色固体状的期望的产物(325mg,42%)
[0345]
uplc-ms(碱性方法,4分钟):rt 2.02分钟,m/z 472.3/474.3[m h] 氯同位素模式
[0346]
1h nmr(400mhz,dmso)δ10.58(s,1h),7.56(s,1h),7.10(d,j=8.6hz,1h),6.93(d,j=2.4hz,1h),6.60(dd,j=8.7,2.4hz,1h),4.26(s,2h),3.72(s,3h),3.55(s,2h),3.45(q,j=6.9hz,2h),2.84(t,j=7.6hz,2h),2.30(s,5h),1.42(s,9h)。
[0347]
步骤2 2-(5-氟吡啶-3-基)-4-{[2-(5-甲氧基-2-甲基-1h-吲哚-3-基)乙基]氨基}-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯
[0348]
根据通用方法b,使用2-氯-4-{[2-(5-甲氧基-2-甲基-1h-吲哚-3-基)乙基]氨基}-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯(325mg)和5-氟吡啶-3-硼酸(97mg)制备,以得到呈褐色固体状的期望的产物(210mg,55%)。
[0349]
uplc-ms(碱性方法,4分钟):rt 1.73分钟,m/z 533.3[m h]
[0350]
19f nmr(376mhz,dmso)δ-127.73。质子去耦
[0351]
1h nmr(400mhz,dmso)δ10.53(s,1h),9.30(d,j=1.8hz,1h),8.65(d,j=2.8hz,1h),8.26(dt,j=10.1,2.4hz,1h),7.26(s,1h),7.09(d,j=8.6hz,1h),6.90(d,j=2.4hz,1h),6.60(dd,j=8.6,2.4hz,1h),4.38(s,2h),3.68(s,3h),3.67-3.56(m,4h),2.93(t,j=7.4hz,2h),2.40(t,j=5.9hz,2h),2.30(s,3h),1.44(s,9h)。
[0352]
步骤3 2-(5-氟吡啶-3-基)-4-{[2-(5-甲氧基-2-甲基-1h-吲哚-3-基)乙基]氨基}-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-鎓氯化物
[0353]
2-(5-氟吡啶-3-基)-4-{[2-(5-甲氧基-2-甲基-1h-吲哚-3-基)乙基]氨基}-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯(210mg,0.39mmol,1.0eq)用含4m hcl的二噁烷(4.0ml,16.0mmol,40.6eq)处理,并且将所得混合物搅拌3小时。将所得悬浮液过滤,并且固体用乙醚洗涤,然后在抽吸下干燥,以得到呈固体状的期望产物(175mg,91%)
[0354]
uplc-ms(4分钟碱性):rt=1.58分钟,m/z=433.2[m h]
[0355]
19f nmr(376mhz,dmso)δ-126.81(d,j=9.7hz)质子去耦
[0356]
1h nmr(400mhz,dmso)δ10.57(s,1h),9.91(s,2h),9.29(d,j=1.7hz,1h),8.75(d,j=2.8hz,1h),8.37-8.28(m,1h),8.02(s,1h),7.08(d,j=8.7hz,1h),6.96(d,j=2.4hz,1h),6.60(dd,j=8.7,2.4hz,1h),4.20(t,j=4.4hz,2h),3.71(s,5h),3.43(d,j=6.5hz,2h),2.95(t,j=7.5hz,2h),2.70(t,j=6.1hz,2h),2.30(s,3h)
[0357]
实例15和162-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h,9h-嘧啶并[4,5-d]氮杂卓-4-胺(实例16)和7-乙基-2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h,9h-嘧啶并[4,5-d]氮杂卓-4-胺(实例15)两者的制备
[0358][0359]
步骤1 7-苄基-2-氯-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h,9h-嘧啶并[4,5-d]氮杂卓-4-胺
[0360]
根据通用方法a,使用2,4-二氯-5,6,8,9-四氢-7h-嘧啶并[4,5-d]氮杂卓-7-羧酸苄酯(131mg)和2-(6-甲氧基-1h-吲哚-3-基)乙-1-胺(80mg)制备,以在使用biotage(telos柱25g,用一定梯度即60至80%etoac的etoac/庚烷洗脱)通过自动柱色谱法进行纯化后得到呈白色固体状的期望的产物(96mg,44%)
[0361]
uplc-ms(碱性,2分钟):rt 1.22分钟,m/z 462.4/464.4[m h] 氯同位素模式。
[0362]
1h nmr(400mhz,dmso)δ10.60(s,1h),7.50(d,j=8.6hz,1h),7.44(t,j=5.5hz,1h),7.34(d,j=4.4hz,4h),7.29-7.24(m,1h),7.01(d,j=2.2hz,1h),6.84(d,j=2.3hz,1h),6.64(dd,j=8.6,2.3hz,1h),3.76(s,3h),3.61(s,2h),3.53(q,j=6.2,5.5hz,2h),
2.88(t,j=7.6hz,2h),2.84-2.79(m,2h),2.70-2.63(m,2h),2.55(d,j=9.2hz,2h)。缺少一个ch2(可能在dmso峰下)。
[0363]
步骤2 7-苄基-2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h,9h-嘧啶并[4,5-d]氮杂卓-4-胺
[0364]
根据通用方法b,使用7-苄基-2-氯-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h,9h-嘧啶并[4,5-d]氮杂卓-4-胺(96mg)和(5-氟吡啶-3-基)硼酸(37mg,1.50eq)制备,以在由mtbe研磨后得到呈白色固体状的期望的产物(51mg,56%)。
[0365]
uplc-ms(碱性方法,2分钟):rt 1.31分钟,m/z 523.4[m h]
[0366]
19f nmr(376mhz,dmso)δ-127.79质子去耦
[0367]
1h nmr(400mhz,dmso)δ10.62(s,1h),9.32(t,j=1.7hz,1h),8.65(d,j=2.9hz,1h),8.32(ddd,j=10.1,2.9,1.6hz,1h),7.43(d,j=8.6hz,1h),7.37-7.32(m,4h),7.30-7.22(m,2h),7.04(d,j=2.2hz,1h),6.84(d,j=2.2hz,1h),6.62(dd,j=8.6,2.3hz,1h),3.76(s,4h),3.64(s,2h),2.98(t,j=7.3hz,4h),2.75(s,2h),2.64-2.58(m,2h)
[0368]
步骤3 2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h,9h-嘧啶并[4,5-d]氮杂卓-4-胺和7-乙基-2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h,9h-嘧啶并[4,5-d]氮杂卓-4-胺
[0369]
在氮气下,将10%钯/碳(20mg)于乙醇(1ml)中的浆液添加到7-苄基-2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h,9h-嘧啶并[4,5-d]氮杂卓-4-胺(51mg)于thf(1ml)中的溶液,并且将所得混合物在1atm下氢化24小时。通过硅藻土塞对催化剂进行过滤,并且用另外的乙醇彻底洗涤。将滤液蒸发至最小体积并且通过制备型hplc(碱性洗脱液)纯化,以得到呈白色固体状的期望的产物2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h,9h-嘧啶并[4,5-d]氮杂卓-4-胺(9.4mg,22.1%)和7-乙基-2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h,9h-嘧啶并[4,5-d]氮杂卓-4-胺(2.8mg,6.2%)
[0370]
2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]-5h,6h,7h,8h,9h-嘧啶并[4,5-d]氮杂卓-4-胺的数据(实例16)
[0371]
uplc-ms(4分钟碱性):rt=1.58分钟,m/z=433.5m h]
[0372]
19f nmr(376mhz,dmso)δ-127.79质子去耦
[0373]
1h nmr(400mhz,dmso)δ10.57(s,1h),9.25(t,j=1.7hz,1h),8.59(d,j=2.9hz,1h),8.25(ddd,j=10.1,2.9,1.6hz,1h),7.37(d,j=8.6hz,1h),7.17(t,j=5.6hz,1h),6.98(d,j=2.2hz,1h),6.77(d,j=2.2hz,1h),6.55(dd,j=8.6,2.3hz,1h),3.68(m,4h),2.89(dd,j=17.1,8.7hz,3h),2.78-2.70(m,2h),2.70-2.62(m,3h),2.51(m,3h)(没有看到一个可交换质子)。
[0374]
7-乙基-2-(5-氟吡啶-3-基)-n-[2-(6-甲氧基-1h-吲哚-3-基)乙基]5h,6h,7h,8h,9h-嘧啶并[4,5-d]氮杂卓-4-胺的数据(实例15)
[0375]
uplc-ms(4分钟碱性):rt=1.86分钟,m/z=461.4[m h]
[0376]
19f nmr(376mhz,dmso)δ-127.76质子去耦
[0377]
1h nmr(400mhz,dmso)δ10.65(s,1h),9.33(t,j=1.7hz,1h),8.66(d,j=2.9hz,1h),8.33(ddd,j=10.1,2.9,1.6hz,1h),7.43(d,j=8.6hz,1h),7.30(t,j=5.7hz,1h),
7.05(d,j=2.2hz,1h),6.84(d,j=2.2hz,1h),6.62(dd,j=8.6,2.3hz,1h),3.78-3.68(m,4h),3.03-2.91(m,4h),2.73(s,2h),2.65-2.57(m,2h),1.03(t,j=7.1hz,3h)
[0378]
实例17 3-(2-{[2-(5-氟吡啶-3-基)-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-4-基]氧基}乙基)-6-甲氧基-1h-吲哚盐酸盐的制备
[0379][0380]
步骤1 2-氯-4-[2-(6-甲氧基-1h-吲哚-3-基)乙氧基]-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯
[0381]
2-(6-甲氧基-1h-吲哚-3-基)乙-1-醇(321mg,1.01eq)于thf(10ml)中的搅拌溶液用氢化钠(86mg,1.30eq,60%分散在矿物油中)在环境温度下处理,并且将所得混合物搅拌10分钟。然后将2,4-二氯-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯(505mg,1.0eq)于thf(10ml)中的溶液在5分钟内以逐滴方式添加到反应容器,并且将所得混合物在环境温度下搅拌2小时。将反应冷却并且蒸发至干燥以得到残余物,将其溶解在etoac中并且用水洗涤。将有机相蒸发至干燥以得到残余物,将其通过柱色谱法(telos 120g盒)纯化,用一定梯度的etoac/庚烷(10至30%etoac)洗脱,以得到呈白色固体状的期望的产物(109mg,14%)。
[0382]
uplc-ms(碱性方法,2分钟):rt 1.26分钟,m/z 459.4/461.4[m h] 氯同位素模式
[0383]
1h nmr(400mhz,dmso)δ10.71-10.63(m,1h),7.47(d,j=8.6hz,1h),7.09(d,j=2.3hz,1h),6.85(d,j=2.2hz,1h),6.66(dd,j=8.6,2.3hz,1h),4.56(t,j=6.9hz,2h),4.42(s,2h),3.76(s,3h),3.57(t,j=5.8hz,2h),3.30(s,2h),3.11(t,j=6.9hz,2h),1.43(s,9h)
[0384]
步骤2 2-(5-氟吡啶-3-基)-4-[2-(6-甲氧基-1h-吲哚-3-基)乙氧基]-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯
[0385]
根据通用方法b,使用2-氯-4-[2-(6-甲氧基-1h-吲哚-3-基)乙氧基]-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯(109mg)和5-氟吡啶-3-硼酸(44mg)制备,以得到呈
5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-鎓氯化物
[0412]
2-(5-氟吡啶-3-基)-4-({2-[2-(三氟甲基)-1h-吲哚-3-基]乙基}氨基)-5h,6h,7h,8h-吡啶并[3,4-d]嘧啶-7-羧酸叔丁酯(30mg,0.054mmol,1.0eq)用含4m hcl的二噁烷(1ml)处理,并且将所得混合物搅拌2小时。形成通过过滤收集的悬浮液。将固体用乙醚洗涤并且在抽吸下干燥,以得到呈固体状的期望的产物(26mg,96%)
[0413]
uplc-ms(碱性方法,4分钟):rt 1.72分钟,m/z 457.2[m h]
[0414]
19f nmr(376mhz,dmso)δ-56.35,-127.60质子去耦
[0415]
1h nmr(400mhz,dmso)δ11.96(s,1h),9.45(s,2h),9.29(t,j=1.7hz,1h),8.71(d,j=2.9hz,1h),8.28(ddd,j=10.0,2.9,1.6hz,1h),7.77(d,j=8.0hz,1h),7.67(t,j=6.0hz,1h),7.43(dt,j=8.3,0.9hz,1h),7.29(ddd,j=8.2,7.0,1.1hz,1h),7.13(ddd,j=8.0,7.0,1.0hz,1h),4.17(s,2h),3.78(q,j=6.8hz,2h),3.45(d,j=6.5hz,2h),3.18(t,j=7.5hz,2h),2.64(q,j=7.3,6.2hz,2h)
[0416]
生物活性
[0417]
在所选生物学测定中对实例进行了两次或更多次测试。数据以pic
50
(-log
10
ic
50
)值的算术平均值报告,其中ic50被定义为产生对激动剂应答的50%抑制的化合物的浓度。在u937测定的情况下,vaf347用作激动剂;在外周血单核细胞(pbmc)测定中,针对pbmc活化后生成的内源性产生的ahr激动剂评估测试化合物的效果。
[0418]
在以下测定中评估了本发明化合物的体外活性:
[0419]
体外测定1:u937细胞中的ahr拮抗(普洛麦格(promega)p450-glo
tm
测定)
[0420]
评估了在u937细胞(衍生自人类组织细胞淋巴瘤的骨髓谱系细胞系)中的ahr拮抗配体在细胞质中结合ahr,并且ahr-配体复合物易位到细胞核并且与ahr核转位子(arnt)形成异二聚体。此复合物在cyp1a1启动子的5
′
上游区域中结合异型生物质应答元件(xre),从而增强cyp1a1表达。随后通过评估萤光素-cee向萤光素的转化来确定cyp1a1活性,所述萤光素进而与萤光素酶反应以产生光。产生的光非量与细胞色素p450活性直接成正比。
[0421]
将ultraculture无血清培养基(龙沙公司(lonza))中的u937细胞以每孔100,000个细胞铺板在圆底96孔组织培养板中。添加七种浓度的测试化合物(最终[dmso]1%)并且温育10分钟,然后添加4.5nm vaf347。然后将板放置在37℃、≥85%湿度、5%co2的培养箱中,持续24小时。吸出上清液后,添加cyp1a1底物萤光素-cee([最终]83μm)并且温育3小时,然后通过添加萤光素检测试剂终止反应,并且在20分钟后读取发光。
[0422]
体外测定2:人外周血单核细胞(pbmc)中的ahr拮抗-抑制白细胞介素-22(il-22)的释放。
[0423]
使用lymphoprep
tm
从人外周血中分离pbmc,并且在含有10%胎牛血清、1%青霉素-链霉素和1%非必需氨基酸的rpmi培养基中稀释至1
×
106个细胞/毫升。随后用每100,000个细胞1μl的cd3/cd28激动剂混合物(人t细胞transact
tm
(美天旎生物技术公司(miltenyi biotec)))活化pbmc,并且然后以每孔100,000个细胞接种在圆底96孔组织培养板中。刺激后一小时,添加七种浓度的每种测试化合物或媒剂(最终[dmso]0.2%)。然后将板放置在37℃、≥85%湿度、5%co2的培养箱中,持续72小时,然后将培养基移除并且储存在-20℃下直到细胞因子分析。根据制造商的说明,使用人il-22duoset elisa(研发系统(r&d systems))测量il-22。
[0424]
结果在表2中示出。
[0425]
实例编号:u937测定(pic
50
)pbmc测定(pic
50
)16.96.956.76.497.76.1107.66.1147.06.2156.86.2166.56.7177.37.6187.76.5
[0426]
体外测定:cyp1a1抑制测定
[0427]
还使用了普洛麦格p450-glo
tm
测定系统对测试化合物的直接cyp1a1抑制活性进行了评估。将七种浓度的测试化合物添加到1/2面积的白色96孔板中。在0.1m磷酸钾缓冲剂中制备cypex cyp1a1细菌小体([最终]0.5pmol)和cyp1a1底物荧光素-cee([最终]30μm),并且在37℃下与测试化合物一起温育5分钟。然后将0.2mm nadph添加到板中并且在37℃下温育10分钟。通过添加荧光素检测试剂终止反应,并且在20分钟后读取发光。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。