一类基于三联苯结构含偶氮苯的芳香胺、
α-二亚胺配体、镍催化剂及其制备方法与应用
技术领域
1.本发明属于催化剂技术领域,具体涉及一类基于三联苯结构含偶氮苯的芳香胺、α-二亚胺配体、镍催化剂及其制备方法与应用,尤其涉及该镍催化剂在催化烯烃聚合制备聚烯烃中的应用。
背景技术:
2.聚烯烃材料由于其本身优异的性能以及相对低廉的价格已经成为合成树脂中产量最大、用途最广的高分子材料。
3.针对于聚烯烃结构的调控,现有技术中最常用的手段是调节配体的电子效应和轴向空间位阻效应。
4.α-二亚胺镍、钯催化剂(j.am.chem.soc.1995,117,6414)具有非常高的活性,从发展到现在已经成为一类应用非常广泛的烯烃聚合催化剂。现有技术中,光响应α-二亚胺偶氮催化剂通过光照条件可以转换调控配体的空间效应与电子效应,实现聚烯烃结构的调控。但是由于光响应α-二亚胺偶氮催化剂的合成难度,已报道的光响应α-二亚胺偶氮催化剂只在金属中心空间对位,而且光响应偶氮单元数通常较少(1~2个),故而现有的光响应α-二亚胺偶氮催化剂没有充分利用偶氮顺反异构的空间位阻变化,因此在某种程度上限制了对聚烯烃结构调控的应用范围。
技术实现要素:
5.有鉴于此,本发明提供一类基于三联苯结构含偶氮苯的芳香胺、α-二亚胺配体、镍催化剂及其制备方法与应用。
6.为实现上述目的,采用以下技术方案:
7.本发明的基于三联苯结构含偶氮苯的芳香胺,结构式如式(i)所示:
[0008][0009]
式(i)中,r2表示h、ch3、c1~c20的烷基、
t
bu(叔丁基)、f、cl、c1~c20 的烷氧基、cf3、no2或n(ch3)2;
[0010]
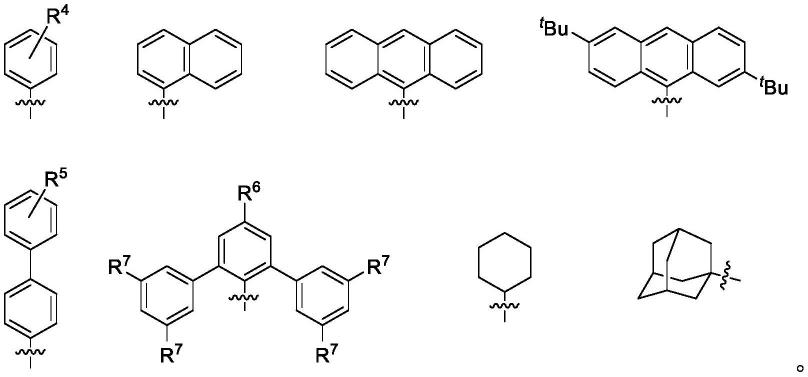
r3表示以下结构中的一种,式中,r4表示h、ch3、
t
bu(叔丁基)、f、cl、 och3、cf3、no2或n(ch3)2,r4位于苯环的邻位、间位或对位;r5表示h、 ch3、
t
bu(叔丁基)、f、cl、och3、cf3、no2或n(ch3)2,r5位于苯环的邻位、间位或对位;r6表示h、f、och3、ch3、cl、cf3、no2或n(ch3)2; r7表示h、ch3、
t
bu(叔丁基)、f、cl、och3、cf3、no2或n(ch3)2;
[0011][0012]
本发明提供一类基于三联苯结构含偶氮苯的α-二亚胺配体,结构式如式(ii) 所示:
[0013][0014][0015]
式(ii)中,r1表示h、c1~c20的烷基,或者表示以下结构中的一种,式中,r8表示h、f、och3、ch3、cl、cf3、no2或n,n-二甲胺基,r8位于苯环的邻位、间位或对位;r9表示f、cl、br、i、och3或ch3;
[0016][0017]
r2表示h、ch3、c1~c20的烷基、
t
bu(叔丁基)、f、cl、c1~c20的烷氧基、cf3、no2或n
(ch3)2;
[0018]
r3表示以下结构中的一种,式中,r4表示h、ch3、
t
bu(叔丁基)、f、cl、 och3、cf3、no2或n(ch3)2,r4位于苯环的邻位、间位或对位;r5表示h、 ch3、
t
bu(叔丁基)、f、cl、och3、cf3、no2或n(ch3)2,r5位于苯环的邻位、间位或对位;r6表示h、f、och3、ch3、cl、cf3、no2或n(ch3)2; r7表示h、ch3、
t
bu(叔丁基)、f、cl、och3、cf3、no2或n(ch3)2。
[0019]
本发明还提供上述基于三联苯结构含偶氮苯的α-二亚胺配体的制备方法,步骤如下:
[0020]
将式(i)所示基于三联苯结构含偶氮苯的芳香胺与式(a)所示二酮按照物质的量比为n:1,n≥2,溶于有机溶剂中,加入催化剂量的催化剂,在回流下搅拌48h以上,冷却至室温,蒸发有机溶剂直至出现固体,将固体析出、过滤、洗涤、真空干燥后,得到式(ii)所示的基于三联苯结构含偶氮苯的α-二亚胺配体(产率在80%以上);
[0021]
所述催化剂为对甲苯磺酸一水合物、甲酸、乙酸中的一种或多种;式(a) 和式(i)的结构分别如下:
[0022][0023]
式(i)中,r2表示h、ch3、c1~c20的烷基、
t
bu(叔丁基)、f、cl、c1~c20 的烷氧基、cf3、no2或n(ch3)2;
[0024]
r3表示以下结构中的一种,式中,r4表示h、ch3、
t
bu(叔丁基)、f、cl、 och3、cf3、no2或n(ch3)2,r4位于苯环的邻位、间位或对位;r5表示h、 ch3、
t
bu(叔丁基)、f、cl、och3、cf3、no2或n(ch3)2,r5位于苯环的邻位、间位或对位;r6表示h、f、och3、ch3、cl、cf3、no2或n(ch3)2; r7表示h、ch3、
t
bu(叔丁基)、f、cl、och3、cf3、no2或n(ch3)2;
[0025][0026]
合成路线为:
[0027][0028]
优选的是,所述有机溶剂为甲苯、二甲苯中的一种或多种的混合。
[0029]
优选的是,所述将固体析出、过滤、洗涤、真空干燥的过程为:加入过量的甲醇或乙醇析出,过滤分离固体,用甲醇或乙醇洗涤三次,并在真空下干燥。
[0030]
优选的是,所述蒸发有机溶剂采用旋蒸蒸发。
[0031]
优选的是,所述回流温度为120℃以上。
[0032]
优选的是,所述催化剂为0.001当量以上。
[0033]
本发明提供一种镍催化剂,结构式如式(iii)或式(iv)所示:
[0034][0035][0036]
式(iii)和式(iv)中,r1表示h、c1~c20的烷基,或者表示以下结构中的一种,式中,r8表示h、f、och3、ch3、cl、cf3、no2或n,n-二甲胺基,r8位于苯环的邻位、间位或对位,r9表示f、cl、br、i、och3或ch3;
[0037]
[0038]
r2表示h、ch3、c1~c20的烷基、
t
bu(叔丁基)、f、cl、c1~c20的烷氧基、cf3、no2或n(ch3)2;
[0039]
r3表示以下结构中的一种,式中,r4表示h、ch3、
t
bu(叔丁基)、f、cl、 och3、cf3、no2或n(ch3)2,r4位于苯环的邻位、间位或对位;r5表示h、 ch3、
t
bu(叔丁基)、f、cl、och3、cf3、no2或n(ch3)2,r5位于苯环的邻位、间位或对位;r6表示h、f、och3、ch3、cl、cf3、no2或n(ch3)2; r7表示h、ch3、
t
bu(叔丁基)、f、cl、och3、cf3、no2或n(ch3)2;
[0040][0041]
本发明还提供上述式(iii)所示的镍催化剂的制备方法,步骤如下:
[0042]
将式(ii)结构的基于三联苯结构含偶氮苯的α-二亚胺配体和(dme)ni (dme=1,2-二甲氧基乙烷)按照物质的量比为1:1溶解于有机溶剂中,在20~50℃下搅拌反应12h以上,真空下蒸发有机溶剂后重结晶,经过滤、洗涤、真空干燥后,得到式(iii)所示的镍催化剂(产率在80%以上)。
[0043]
合成路线为:
[0044][0045]
优选的是,所述有机溶剂为二氯甲烷或氯仿。
[0046]
优选的是,所述重结晶采用的混合液为以下混合液中的一种:正己烷和二氯甲烷的混合液、正己烷和氯仿的混合液、乙醚和氯仿的混合液、乙醚和二氯甲烷的混合液。
[0047]
优选的是,所述洗涤为用正己烷或乙醚洗涤三次。
[0048]
本发明还提供上述式(iv)所示的镍催化剂的制备方法,步骤如下:
[0049]
将式(ii)结构的基于三联苯结构含偶氮苯的α-二亚胺配体、ni(acac)
2 (acac=乙酰丙酮)和[ph3c][b(c6f5)4按照物质的量比为1:1:1溶解于有机溶剂中,室温下搅拌反应
12h以上,真空下蒸发有机溶剂,经洗涤、过滤、真空干燥后,得到式(iv)所示的镍催化剂(产率在70%以上)。
[0050]
合成路线为:
[0051][0052]
优选的是,所述洗涤、过滤为用正己烷、乙醚依次洗涤后过滤,重复三次。
[0053]
本发明还提供上述镍催化剂在催化乙烯制备聚烯烃中的应用。
[0054]
在惰性气氛下,将助催化剂溶于有机溶剂中,得到溶液a,将镍催化剂溶于有机溶剂,得到溶液b,在黑暗环境或者10~400nm波长的紫外光照射环境下,搅拌下,将溶液b加入溶液a中,控制乙烯的压力为1~20atm,反应温度为 0~120℃,反应5~120min,反应结束,加入含有盐酸的醇溶液淬灭聚合反应,过滤,烘干至恒重,得到聚烯烃。
[0055]
更优选的是,所述有机溶剂为二氯甲烷、氯仿或己烷;所述助催化剂为mao、mmao、easc或alet2cl;所述助催化剂和镍催化剂的物质的量比为大于等于 100:1,尤其优选的是大于等于500:1;所述含有盐酸的醇溶液为盐酸浓度为5wt%以上的甲醇溶液或盐酸浓度为5wt%以上的乙醇溶液。
[0056]
更优选的是,所述聚烯烃为聚乙烯。
[0057]
更优选的是,所述紫外光的波长为300~400nm。
[0058]
在本发明中所使用的术语,一般具有本领域普通技术人员通常理解的含义,除非
另有说明。本发明定义室温为20-25℃。
[0059]
本发明的原理为:偶氮单元在不同光照条件下具有可逆顺反异构的特征(黑暗下或自然环境下反式构象、紫外光照射下顺式构象),故可利用光来调控烯烃聚合反应,得到不同微观结构和理化性能的聚合物。本发明的镍催化剂,与现有技术中通过调控取代基体积来改变轴向位阻不同,将多个偶氮苯光响应单元置于金属中心的轴向位置,创造性地将偶氮苯光响应作用、位阻效应、电子效应三者相结合,进一步拓展了镍催化剂的应用范围。这类镍催化剂在调控烯烃聚合方面具有非常显著的效果,常温常压下可以在很宽的范围内调控催化活性、聚合物分子量、支化度、支化类型。
[0060]
与现有技术相比,本发明的有益效果为:
[0061]
本发明提供了一类基于三联苯结构含偶氮苯的芳香胺,并进一步合成了相应的α-二亚胺配体及镍催化剂。经试验验证,在常温常压,该镍催化剂在烯烃聚合中有显著的调控效果,具体表现为:
[0062]
1、该镍催化剂可在一定波长光照条件(10~400nm)下改变自身空间结构,达到通过同时调控位阻效应及电子效应调节聚合物性能的目的;
[0063]
2、该镍催化剂经过一定波长(10~400nm)光照后,可在催化活性(在有活性的基础上,差值最高可达10.2倍)、聚合物分子量(mw差值最高8.5倍)、支化密度(差值最高可达4.7倍)、支化类型(长链支化最高可相差15%)上实现宽范围调控;
[0064]
3、该镍催化剂在光调控烯烃聚合过程中,对于聚合活性的影响极大,甚至可能导致有无聚合物的差别。
附图说明
[0065]
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其它的附图。
[0066]
图1为本发明实施例1中的镍催化剂的单晶衍射图;
[0067]
图2为本发明实施例1中的镍催化剂的核磁共振氢谱图;
[0068]
图3为本发明实施例81中的聚合物的核磁共振氢谱图。
具体实施方式
[0069]
为了使本领域的技术人员更好地理解本发明的技术方案,下面将结合实施例对本发明作进一步的详细介绍。
[0070]
在以下实施例中,未详细描述的各种过程和方法是本领域中公知的常规方法。下述实施例中所用的材料、试剂、装置、仪器、设备等,如无特殊说明,均可从商业途径获得。
[0071]
实施例1
[0072]
步骤一、4-甲基-2,6-双(4,4,5,5-四甲基-1,3,2-二氧杂硼烷)苯胺的制备
[0073][0074]
惰性气氛下(氮气),将2,6-二溴-4-甲基苯胺(0.50g,1.9mmol),双(频哪醇)二硼烷(1.44g,5.7mmol),乙酸钾(0.55g,5.6mmol)和[1,1'-双(二苯基膦基)二茂铁]二氯化钯(0.028g)溶解在二甲基亚砜中(20ml),然后在80℃下搅拌24h以上。待反应完成后,将黑红色液体混合物倒入冰水(50ml) 中并过滤沉淀物。将沉淀物重新溶解在二氯甲烷中,用水洗涤有机层,分离有机层并用无水硫酸钠干燥。将固体过滤后,有机相真空旋转蒸发浓缩。通过快速柱色谱法(硅胶;pe/ch2cl2=3∶1)纯化所得残余物,然后使用ch2cl2/ch3oh 重结晶,得到0.40g(60%收率)黄白色晶状产物。
[0075]
步骤二、4-甲基-2,6-二[(4-偶氮苯)苯基]苯胺的制备
[0076][0077]
在氮气气氛的室温下,将4-甲基-2,6-双(4,4,5,5-四甲基-1,3,2-二氧杂硼烷) 苯胺(13.9mmol),4-碘偶氮苯(34.8mmol),四三苯基磷钯(0.8mmol),碳酸钠水溶液(2mol/l),溶解在乙醇与甲苯的混合溶液中(乙醇20ml,甲苯80ml)。然后将该悬浮液在剧烈搅拌下加热至90℃,反应24h。待反应完成后,将悬浊液用二氯甲烷萃取,用水洗涤有机层,并用无水硫酸钠干燥有机层。过滤固体后,将有机相旋转蒸发浓缩。然后将所得残余物通过柱色谱法(硅胶;pe/ch2cl2= 1∶1)纯化,得到橙黄色固体产物(94.8%)。
[0078]
步骤三、双-[2,6-二(4-偶氮苯)苯基-4-甲基苯基)丁二酮-1,2-二亚胺的制备
[0079]
[0080]
将4-甲基-2,6-二[(4-偶氮苯)苯基]苯胺(3.4mmol),2,3-丁二酮(1.6mmol) 和催化量的对甲苯磺酸一水合物(10mg)溶解在50ml甲苯中,该混合溶液在130℃下回流反应48h。反应完成后,恢复至室温,旋蒸蒸发浓缩溶剂,加入100ml 无水乙醇析出黄色固体,过滤,用无水乙醇洗涤三次并在真空下干燥得到黄色固体产物(47.4%收率)。
[0081]
步骤四、[(men^n)nibr2的制备
[0082][0083]
将双-[2,6-二(4-蒽基)苯基-4-甲基苯基)丁二酮-1,2-二亚胺(0.35mmol), (dme)nibr2(0.35mmol)溶解在50ml二氯甲烷中,在25℃下搅拌反应24h,得到深红色液体,真空蒸发溶剂后得到黄色固体,加入正己烷洗涤三次,过滤,最后用乙醚洗涤三次黄色固体,真空下抽干,得到镍催化剂(82.6%收率)。
[0084]
步骤五、[(men^n)ni(acac)][b(c6f5)4]的制备
[0085]
[0086]
将双-[2,6-二(4-偶氮苯)苯基-4-甲基苯基)丁二酮-1,2-二亚胺(0.15mmol),二乙酰丙酮镍(0.15mmol),大硼烷[(c6h5)3][b(c6f5)4](0.15mmol)溶解在25ml二氯甲烷中,在25℃下搅拌反应24h,得到深红色液体,真空蒸发溶剂后得到红色固体,加入正己烷洗涤三次,过滤,最后用乙醚洗涤三次红色固体,真空下抽干,得到镍催化剂(86.9%收率)。
[0087]
对实施例1步骤一、步骤二、步骤三、步骤四和步骤五的产物分别进行核磁或质谱检测,结果如下:步骤一的黄白色晶状产物:1h nmr(500mhz,298k, cdcl3,2.50ppm):δ=7.51(s,2h,aryl-h),5.87(s,2h,nh2),2.18(s,3h, ch3),1.32(s,ch3,24h)。步骤二的橙黄色固体产物:1h nmr(500mhz,298 k,cdcl3,7.26ppm):δ=8.03-8.01(d,4h,aryl-h),7.96-7.94(d,4h,aryl-h), δ=7.71-7.69(d,4h,aryl-h),δ=7.56-7.47(m,6h,aryl-h),7.05(s,2h,aryl-h), 3.83(s,2h,-nh2),2.36(s,3h,aryl-ch3)ppm.步骤三的黄色固体产物:1h nmr(500mhz,298k,cdcl3,7.26ppm):δ=7.95-7.93(m,8h,aryl-h), 7.83-7.81(d,8h,aryl-h),7.48-7.43(m,8h,aryl-h),7.32-7.30(d,8h,aryl-h), 7.16(s,4h,aryl-h),2.39(s,6h,aryl-me),1.44(s,6h,n=c-me)ppm.
13
c{1h} nmr(125mhz,298k,cdcl3,77.16ppm):δ=168.16(n=c-me),152.91, 151.37,143.64,142.98,133.96,131.02,130.95,130.62,129.92,129.14,123.12, 122.68,20.92(aryl-me),17.15(n=c-me)ppm.步骤四的镍催化剂: maldi-tof-ms(m/z):1060.0[m-2br]
,1001.3[m-2br-ni]
.步骤五的镍催化剂:1h nmr(500mhz,298k,cdcl3,7.26ppm):δ=8.14-7.98(d,16h,aryl-h), 7.51-7.37(m,20h,aryl-h),7.16(s,4h,aryl-h),5.42(s,1h,acac-ch),2.37 (s,6h,aryl-me),1.63(s,12h,acac-me),1.56(s,6h,n=c-me)ppm.
13
c{1h} nmr(125mhz,298k,cdcl3,77.16ppm):186.72(me-c=o),176.42 (n=c-me),152.63,152.43,141.20,140.29,136.25,134.07,131.97,131.91,129.41, 129.38,129.43,123.89,123.30,102.25,24.80(me-c=o),21.15(aryl-me),20.72 (n=c-me).
19
f nmr(500mhz,298k,cdcl3):δ=-132.56,-163.14,-166.85ppm.
[0088]
实施例2~18
[0089]
步骤一-步骤五与实施例1相同,除了表1中的变量,其他条件均无变化。表1实施例2~18的步骤一和步骤二合成反应物c(反应温度:90℃,反应时间:24h)
[0090]
[0091]
[0092][0093]
实施例19~40
[0094]
首先将与高压气体管线连接的350ml玻璃压力反应器在90℃下真空干燥至少1h,然后用锡箔纸包裹反应器置于黑暗环境。然后将反应器调节至20℃,在惰性气氛下将98ml二氯甲烷和500μmol的mao加入到反应器中,然后将1 μmol的ni催化剂溶解在2ml二氯甲烷(或氯仿)中通过注射器注入到聚合体系中。在快速搅拌下(600转以上),通入乙烯并保持在1bar。30min后,排空压力反应器,加入大量酸性甲醇(或乙醇)(5%以上的盐酸醇溶液)溶液淬灭聚合反应,过滤聚合物,并在真空烘箱中干燥至恒重。
[0095]
其中,镍催化剂的结构通式如式(iv)所示,r2=ch3,r3=ph,r4=h,r1、 r8、r9见表2。
[0096]
表2实施例19~40室温常压下不同镍催化剂(r1、r8、r9)对乙烯聚合的影响
[0097]
[0098]
[0099][0100]
表2中,活性:以106g mol-1
h-1
为单位。mw、mw/mn:分别为重均分子量、聚合物分散性指数,150℃下,在1,2,4-三氯苯中通过gpc测定,相对于聚苯乙烯标准物。支化度=每1000个碳支化的个数,由核磁共振氢谱测定。所有数据至少是基于两条平行试验得出的结果(除非另有说明)。
[0101]
从表2可以看出,当控制催化剂取代基r2、r3、r4不变时,改变取代基r1、 r8、r9时,在同等的聚合条件下(时间、温度、压力、助催化剂浓度一致),当时,聚合物分子量最高为151.2万。当时,得到最高活性(3.12
×
106g mol-1
h-1
)。其中,当r8为供电子基团(ch3、och3、 n,n-二甲胺基)相比于其为吸电子基团(f、cl、cf3、no2)拥有更高的活性、分子量。当改变r8的位置时,如果r8处于苯基的邻位,该催化剂能得到最高的的分子量,如果r8处于苯基的对位,该催化剂能得到的聚合活性。当r9的位阻越大时,得到的聚合物具有更高的活性、更高的分子量(i》br》cl》f)。其中,除去当
时,同等条件下紫外光照射环境有聚合物产生外(活性0.48
×
106g mol-1
h-1
,mw26.9
×
104g mol-1
,支化度26),其余催化剂均未表现出明显聚合行为。
[0102]
实施例41~68
[0103]
首先将与高压气体管线连接的350ml玻璃压力反应器在90℃下真空干燥至少1h,然后置于不同光照环境下,具体为用锡箔纸包裹反应器置于黑暗环境或者置于紫外光(10~400nm)环境照射。然后将反应器调节至20℃,在惰性气氛下将98ml二氯甲烷和500μmol的mao加入到反应器中,然后将1μmol的ni 催化剂溶解在2ml二氯甲烷(或氯仿)中通过注射器注入到聚合体系中。在快速搅拌下(600转以上),通入乙烯并保持在1bar。30min后,排空压力反应器,加入大量酸性甲醇(或乙醇)(5%以上的盐酸醇溶液)溶液淬灭聚合反应,过滤聚合物,并在真空烘箱中干燥至恒重。
[0104]
其中,镍催化剂的结构通式如式(iv)所示,r2=ch3, r3、r4、r5、r6、r7见表3。
[0105]
表3实施例41~68室温常压下不同镍催化剂(r3、r4、r5、r6、r7)对乙烯聚合的影响
[0106]
[0107][0108]
表3中,活性:以106g mol-1
h-1
为单位。mw、mw/mn:分别为重均分子量、聚合物分散性指数,150℃下,在1,2,4-三氯苯中通过gpc测定,相对于聚苯乙烯标准物。支化度=每1000个碳支化的个数,由核磁共振氢谱测定。所有数据至少是基于两条平行试验得出的结果(除非另有说明)。
[0109]
从表3可以看出,当控制催化剂取代基r1、r2不变时,改变取代基r3、r4、 r5和光照条件时,在同等的聚合条件下(时间、温度、压力、助催化剂浓度一致),光照下催化剂的活性远
时,在同等的聚合条件下(时间、温度、压力、助催化剂浓度一致),此外,吸电子基团(f、cl、cf3、no2) 会导致聚合物分子量及活性下降,而供电子基团是有利于提高聚合物活性及分子量的(
t
bu、och3、n(ch3)2)。
[0118]
实施例78~96
[0119]
首先将与高压气体管线连接的350ml玻璃压力反应器在90℃下真空干燥至少1h,然后置于不同光照环境下,具体为用锡箔纸包裹反应器置于黑暗环境或者置于紫外光(10~400nm)环境照射。然后将反应器调节至0~40℃,在惰性气氛下将98ml二氯甲烷和500μmol的mao加入到反应器中,然后将1μmol的 ni催化剂溶解在2ml二氯甲烷(或氯仿)中通过注射器注入到聚合体系中。在快速搅拌下(600转以上),通入乙烯并保持在1bar。30~240min后,排空压力反应器,加入大量酸性甲醇(或乙醇)(5%以上的盐酸醇溶液)溶液淬灭聚合反应,过滤聚合物,并在真空烘箱中干燥至恒重。
[0120]
其中,镍催化剂的结构通式如式(iv)所示,r3=ph,r4=h,反应条件见表5。
[0121]
表5实施例78~96不同反应条件对不同α-二亚胺镍催化剂催化乙烯聚合的影响
[0122][0123][0124]
表5中,以106g mol-1
h-1
为单位。mw、mw/mn:分别为重均分子量、聚合物分散性指数,150℃下,在1,2,4-三氯苯中通过gpc测定,相对于聚苯乙烯标准物。支化度=每1000个碳支化的个数,由核磁共振氢谱测定。所有数据至少是基于两条平行试验得出的结果(除非另有说明)。
[0125]
从表5可以看出,当保持压力不变(1bar)、温度不变(30℃),聚合物的产率和分子量随着时间的变大而逐渐变大;当保持压力不变(1bar)、时间不变 (30min),r1=ch3二溴镍
催化剂产生聚合物的活性和分子量随着温度的升高而降低,支化度随着温度的升高而明显升高;r1=ch3乙酰丙酮镍催化剂产生聚合物的活性和分子量随着温度的升高而降低,支化度随着温度的升高而明显升高;乙酰丙酮镍催化剂产生聚合物的活性和分子量随着温度的升高而降低,支化度随着温度的升高而明显升高。在同等的聚合条件下(时间、温度、压力、助催化剂浓度一致),聚合物的活性随光照条件改变(从黑暗环境到10~400nm紫外光照射)活性大幅度下降,分子量降低,分子量分布变宽,支化度增加。当使用r1=ch3二溴镍催化剂时,40℃下活性最高差距可达10.23 倍。当使用r1=ch3二溴镍催化剂时,20℃下支化度最高差距可达4.71倍。
[0126]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施例的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有实施例予以穷举。而由此所引申出的显而易见的变化或变动仍处于本发明创造的保护范围之中。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。